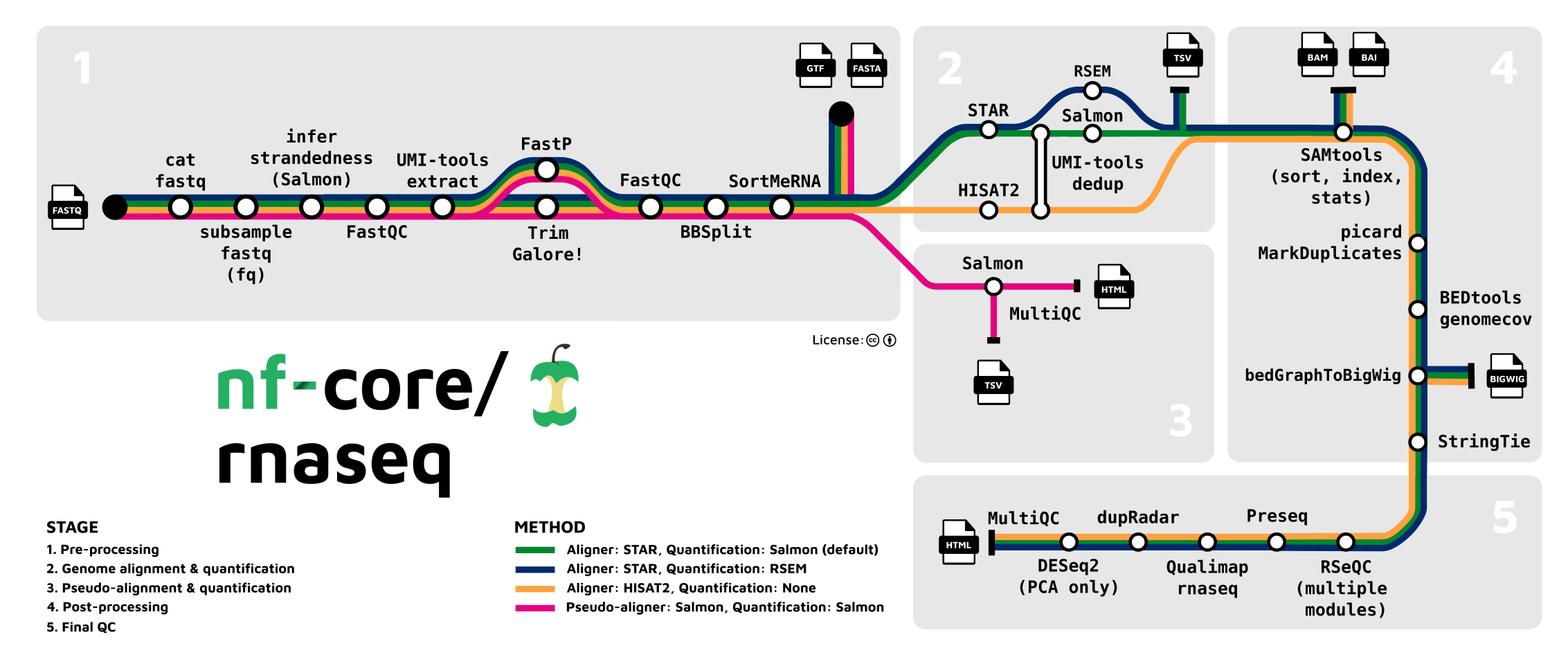
1. 前言
拿到转录组的测序数据,怎么得到基因组中每个基因的表达量呢?正常要经历“指控-比对-定量-标准化”流程,才能得到表达量。而nf-core/rnaseq就是帮助我们一条命令完成上述步骤,直接得到基因表达量的一个基于Nextflow写的pipline。
2. 下载
nf-core类似于一个工具箱,下面会有很多工具,rnaseq就是其中一个,所以要先下载nf-core。
mamba creaye -n nf-core
mamba install bioconda::nf-core
接下来,通过nf-core下载rnaseq
nf-core download nf-core/rnaseq
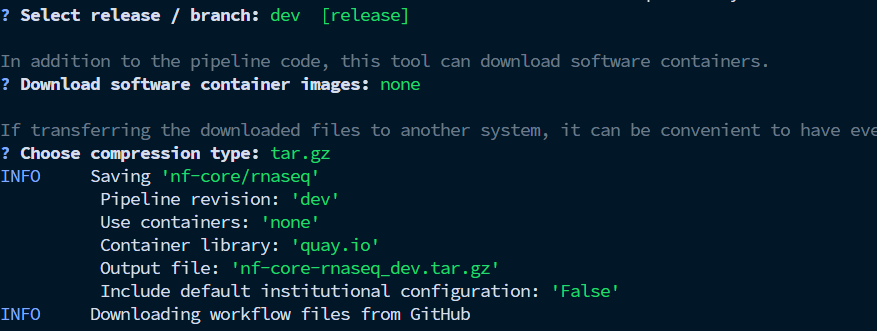
依次选择,dev,none,tar.gz,完成后会出现了一个“nf-core-rnaseq_dev”,里面就是nf-core/rnaseq软件。
3. 运行
nextflow run ~/Software/nf-core/nf-core-rnaseq_dev/dev/ --input sample.csv --gff ../Anno/Final_result/Prunus_pedunculata_Chr.gff --fasta ../Anno/Final_result/Prunus_pedunculata_Chr.fasta --outdir RNA-seq_seticuspe -profile docker
# --input sample.csv: 指定输入的样本信息文件(samplesheet)。这个 CSV 文件通常包含多列,如样本ID、FASTQ文件的路径(单端或双端 R1/R2)、分组信息等,流程会根据这个文件来处理每个样本。
# --gff ../Anno/Final_result/Prunus_pedunculata_Chr.gff: 指定基因组注释文件,格式为 GFF。这个文件包含了基因、转录本、外显子等在基因组上的位置信息,是进行基因表达定量所必需的。
# --fasta ../Anno/Final_result/Prunus_pedunculata_Chr.fasta: 指定参考基因组的序列文件,格式为 FASTA。RNA-seq 的读长(reads)将会比对到这个参考基因组上。
# --outdir RNA-seq_seticuspe: 指定所有分析结果的输出目录。流程运行结束后,所有的报告、比对文件、定量结果等都会保存在这个名为 RNA-seq_seticuspe 的文件夹中。
# -profile docker: 指定运行所使用的配置 profile。docker 是一个预设的 profile,它告诉 Nextflow 使用 Docker 容器来运行流程中的每一个分析步骤(如 STAR 比对, Salmon
定量等)。这样做的好处是,所有软件及其依赖都打包在容器中,确保了分析环境的一致性和可重复性,避免了手动安装大量软件的麻烦。
这里要强调的是,要手动整理一个sample.csv文件,记录了样本的测序文件的路径和信息。
sample,fastq_1,fastq_2,strandedness
SRR18362927,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR18362927_1.fastq.gz,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR18362927_2.fastq.gz,auto
SRR18362928,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR18362928_1.fastq.gz,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR18362928_2.fastq.gz,auto
SRR18362929,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR18362929_1.fastq.gz,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR18362929_2.fastq.gz,auto
SRR30835667,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR30835667_1.fastq.gz,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR30835667_2.fastq.gz,auto
SRR30835668,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR30835668_1.fastq.gz,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR30835668_2.fastq.gz,auto
SRR30835669,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR30835669_1.fastq.gz,/home/majunpeng/sda2/Pangenome_Catus/Hpchic_readjujst/Straned/Catus/Total_vcf/SV_RNA-seq/RNA-seq/Chrysanthemum_indicum/SRR30835669_2.fastq.gz,auto
4. 输出结果
输出6个文件夹,最后需要的定量结果位于star_salmon文件夹下的“salmon.merged.gene_tpm.tsv”和”salmon.merged.transcript_tpm.tsv“分别是基于基因和mRNA的定量结果。