引言
Minimap2 是目前广泛应用于基因组比对领域的一个高效工具,特别适用于长读长数据的比对任务。凭借其出色的性能,Minimap2已成为多种基因组学和转录组学研究中的标准工具。然而,在处理大规模基因组数据时,Minimap2的比对速度往往成为瓶颈,尤其是在高通量数据量激增的情况下,处理时间可能会变得非常缓慢,从而影响整体工作效率。
为了应对这一挑战,mm2-plus应运而生。作为Minimap2的加速版,mm2-plus通过优化算法和计算流程,显著提高了比对速度,同时保持了高精度的比对结果。无论是大规模基因组比对,还是高复杂度的测序数据处理,mm2-plus都能为用户提供更加高效、便捷的解决方案,是基因组比对任务的理想选择。
mm2-plus
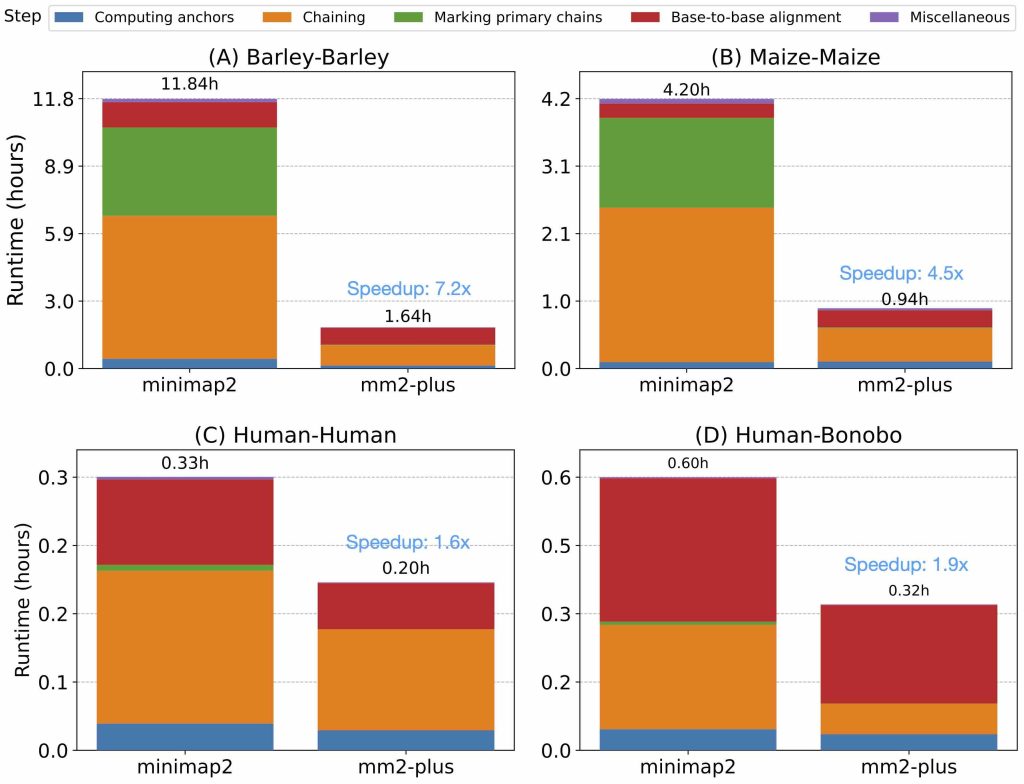
mm2-plus作为minimap2的加速版,比对速度提升的同时,也保证比对结果和minimap2基本一样,因此基本可以算是minimap2的上位替代。文件中说明,mm2plus的加速效率根据基因组的复杂程度不同,可以实现1.6-7.2倍的加速效率。
安装
mamba install bioconda::mm2plus
比对
mm2-plus的命令和minimap2是完全一样的,只需要把minimap2替换为mm2plus即可。
# minimap2
minimap2 -ax asm5 -t 8 --eqx Prunus_pedunculata_Chr_g4.fasta Prunus_pedunculata_Chr_g5.fasta > minimap2.paf
# mm2plus
mm2plus -ax asm5 -t 20 --eqx Prunus_pedunculata_Chr_g4.fasta Prunus_pedunculata_Chr_g5.fasta > mm2plus.paf
我用来测试的基因组大小是250M,都使用20个cpu的情况下,minimap2的比对时间为6分30秒,mm2plus的比对时间为5分16秒,提升大约20%。
接下来使用一个1.5G的植物基因组同时使用minimap2和mm2plus进行全基因组比对,测试加速效果
# minimap2
minimap2 -ax asm5 -t 48 --eqx Prunus_pedunculata_Chr_48.fasta Prunus_pedunculata_Chr_48.fasta > minimap2.paf
# mm2plus
mm2plus -ax asm5 -t 48 --eqx Prunus_pedunculata_Chr_48.fasta Prunus_pedunculata_Chr_48.fasta > mm2plus.paf
minimap2的运行时间为14分16秒,mm2plus的运行时间为8分36秒,速度提升接近1倍。
最后使用一个基因组大小为3G的基因组来进行最后的测试
# minimap2
minimap2 -ax asm5 -t 48 --eqx Chrysanthemum_rhombifolium_Hap_A.fasta Chrysanthemum_rhombifolium_Hap_A.fasta > minimap2.paf
# mm2plus
mm2plus -ax asm5 -t 48 --eqx Chrysanthemum_rhombifolium_Hap_A.fasta Chrysanthemum_rhombifolium_Hap_A.fasta > mm2plus.paf
这时候出现了问题,minimap2可以正常运行,单数mm2plus出现了(core dumped)错误,应该是内存直接爆了。
结语
总体来看mm2plus还是足够优秀的,可以大幅度提升minimap2算法的比对速度,但是提升速度的同时也需要消耗更多的内存,因此对mm2plus对内存的需求更高。