前言
在上一篇文章中,我们系统介绍了 ANNEVO 与 Helixer 两款基于深度学习的从头基因预测软件的环境配置、模型调用及实际使用流程。整体而言,这类方法在复杂基因结构识别、弱证据区域预测以及大规模基因组注释中展现出了显著优势,已逐渐成为基因组注释流程中的重要组成部分。
然而,在实际应用中也可以明显感受到,这两款软件存在一个共同的瓶颈:官方提供的预训练模型在分类尺度上过于粗粒度。以 Helixer 为例,目前所有陆生植物均统一使用同一个 land_plant 模型。显然,陆生植物涵盖了从苔藓、蕨类到裸子植物和被子植物等极其庞大的系统发育跨度,不同类群在基因密度、内含子长度、剪接模式以及转座子背景等方面均存在显著差异。仅依赖一个通用模型,难以在所有植物类群中同时获得最优预测效果,在某些特定类群中出现预测偏差几乎是不可避免的。
因此,基于特定类群的高质量基因组与注释结果,对现有模型进行针对性的微调(fine-tuning),构建“类群感知”的注释模型,具有重要的现实意义。这不仅有助于提升目标类群中新基因组的注释准确性和一致性,也为在更大系统发育尺度上构建分层、可迁移的注释模型提供了可行路径。
构建训练数据集
准备一个分类单元下所有已发表基因组的注释结果,这些基因组最好是高质量的染色体水平以上的,并且注释的时候整合多方证据得到的高质量注释。最后通过GeenuFF对注释结果进行过滤,删除存在明显错误的注释并储存在sqlite数据库中。GeenuFF主要过滤的错误有起始密码子缺失或位置不对、终止密码子缺失或提前出现、 5′ 端相位错误、3′ 端相位不匹配、外显子重叠和内含子太短 。
下载GeenuFF
git clone https://github.com/weberlab-hhu/GeenuFF.git
cd GeenuFF
pip install -r requirements.txt
pip install .
GeenuFF对注释结果进行过滤,并转换为sqlite
python3 import2geenuff.py --gff3 Prunus_mongolica_Chr.gff \
--fasta Prunus_mongolica_Chr.genome_trimmed.fasta \
--db-path Prunus_mongolica.sqlite3 \
--log-file Prunus_mongolica.log \
--species Prunus_mongolica
# --fasta 基因组文件
# --gff3 基因组注释文件
# --db-path 输出qlite文件
# --log-file log文件
# --species 物种
sqlite转换为Helixer训练数据要求的h5文件
apptainer run --nv ~/helixer-docker_latest.sif geenuff2h5.py \
--input-db-path Prunus_mongolica.sqlite3 \
--h5-output-path Prunus_mongolica.h5
将每个用作训练和验证的物种的基因组对应的h5文件整理到一个文件夹中,训练数据集的h5文件名并且符合training_data*格式,验证的数据集h5文件名必须符合validation_data*,不然会报错。
训练模型
apptainer run --nv ~/helixer-docker_latest.sif HybridModel.py \
--data-dir train \
--save-model-path best_helixer_model.h5 \
--epochs 50 \
--predict-phase \
--load-model-path /home/majunpeng/.local/share/Helixer/models/land_plant/land_plant_v0.3_a_0080.h5\
--resume-training
# --data-dir 训练和验证数据集的路径
# --save-model-path 训练后的模型
# --epochs 50
# --load-model-path 加载的模型
20260201补充:不知道配置了什么,现在要加上–env PYTHONNOUSERSITE=1才能成功运行。
随后会对模型进行训练,并统计训练后模型的一些指标,衡量训练模型预测基因的准确度。
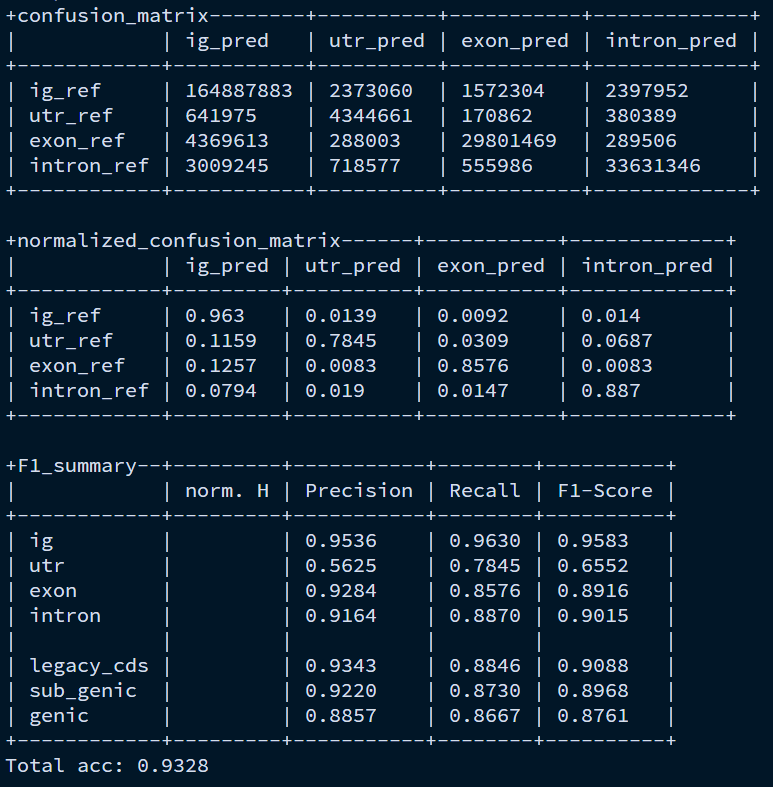
训练模型结果解读
首先来看下基因组四个区域的含义:
ig:基因间隔区
utr:UTR区
exon:外显子
intron:内含子
Confusion Matrix:统计每个碱基的分类结果,每一个碱基,都被模型预测成 4 类之一,同时,该碱基在参考注释中也属于这 4 类之一。因此,混淆矩阵就是真实类别(行) × 预测类别(列) 的碱基数量统计。
比如| exon_ref | 5014284 | 2002047 | 2001614 | 134915 |
表示有 5,014,284 bp 的真实 exon被预测成 IG;有 2,001,614 bp被预测成 exon(正确)
Normalized Confusion Matrix:对Confusion Matrix按行归一化(row-normalized)。
F1_summary:统计了每个区域的召回率、准确率和F1-score。下面三个指标什么意思没搞懂。
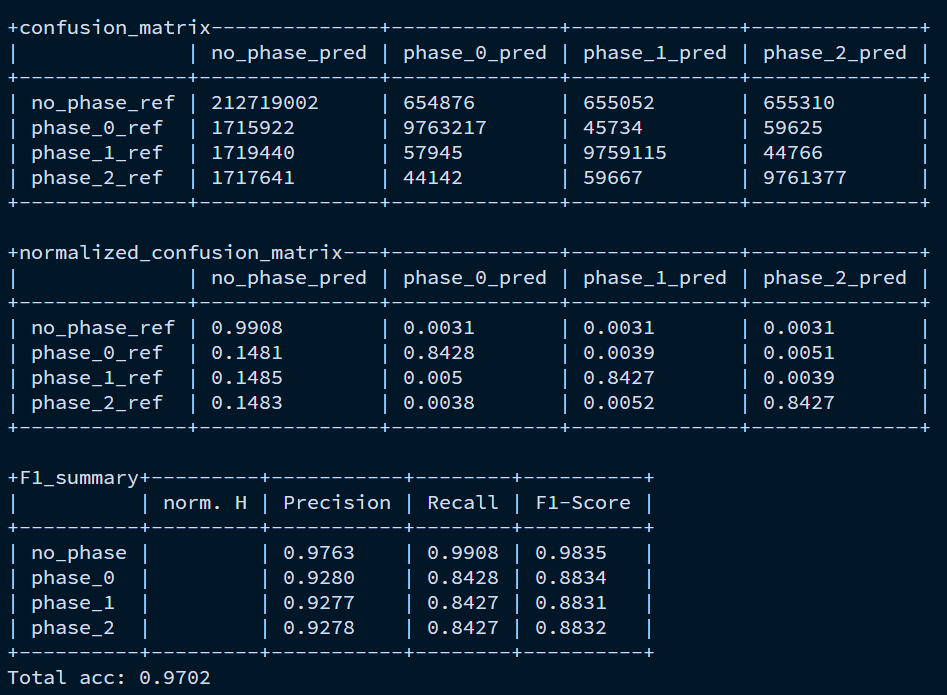
和上面统计的指标类似,只不过关注的是phase。phase 描述的是:某一个碱基在 CDS 密码子(三联体)中的位置。因此这里统计的的是CDS内部每个密码子的位置预测的准确率,关注的是CDS内部有没有出现移码等错误。
| phase | 含义 |
|---|---|
| phase 0 | 密码子的第 1 个碱基 |
| phase 1 | 密码子的第 2 个碱基 |
| phase 2 | 密码子的第 3 个碱基 |
| no_phase | 不在 CDS 中 |
综合一句话就是,第一个表统计的是,基因区域预测的准不准,第二个表统计的是基因内部CDS预测的对不对。
结语
经过我自己测试下来,针对某一个类群进行单独的模型训练,得到一个针对单个类群的注释模型是真重要的,对注释的结果有良好的改善。后续准备针对这个方面单独发表一篇文章,欢迎有基因组基础和机器学习基础老师和同学联系我,一起合做研究。