0.前言
作为一个基因组学研究人员,怎么可以不会GWAS呢?最近接了一个GWAS的项目,正好借着这个机会边做边学GWAS。(加笔:耗时半个月,大致学完了GWAS的流程,有一个初步的理解。虽然GWAS是早就出现的技术,但是放到今天确实不过时,通过将SNP等变异和表型数据关联,得到相关的位点,确实能比较精准的得到和关键性状相关的基因。但是!如果想得到准确的结果,分析的流程要非常标准,比如表型的采集和记录,大多人会低估这一步,但是我认为是比较重要的。如果错误的表型很有可能会导致不能找到准确的SNP位点,或者根本找不到SNP位点,分析出花来都没用。另外GWAS分析的主流软件和模型回对结果有较明显的结果,所以如果结果不好,或者找不到P值明显的点,可以换模型试一下。最后,初学者可能觉得找到SNP位点就结束了,但是很有可能SNP位点压根不在基因中,这时候就需要通过其他软件来计算连锁不平衡,来找到和SNP位点具有不平衡的基因,作为候选基因。)
正式开始之前,先放一些和VCF文件处理和过滤相关的命令。
0.1 统计并可视化VCF文件中SNP位点的缺失率
# Stat
plink --vcf Rename_snps.f4.withid.75samples.RNAseqID.vcf --missing --out demo
# Plot
0.2 对基因型位点进行过滤
plink --vcf demo.vcf --out demo.final_clean --recode vcf-iid --remove demo.fail.samples --make-bed --geno 0.1 --maf 0.05 --biallelic-only strict --hwe 1e-6 -set-missing-var-ids @:# --allow-extra-chr
0.3 对基因型位点进行填充
beagle -Xmx4g -Djava.io.tmpdir=./TMP gt=./demo.final_clean.vcf out=demo.impute impute=true window=10 nthreads=4
0.4 检查表型数据的正态性
Rscript ../script/phenotype_check.R ../data/demo_traits.txt traits_check
0.4 计算亲缘关系矩阵
gcta --bfile ../00.prepare/demo --make-grm-gz --make-grm-alg 0 --out gcta_kinship
## 替换sample name ; 转成对角矩阵
gzip -d gcta_kinship.grm.gz
awk -v OFS="\t" 'NR==FNR{ A[ NR ] = $1}; NR>FNR{ if( $1 in A && $2 in A){ $1 = A[$1]; $2 = A[$2] ; print $0}}' gcta_kinship.grm.id gcta_kinship.grm > gcta_kinship.grm.addname.pairwise
awk -v OFS="\t" '{if(NR==FNR){ A[NR] = $1; max=NR } else { B[$1][$2] = $4; B[$2][$1] = $4 } } END { for( i=1; i<=max; i++){ printf A[i] ; for(j=1; j<=max; j++){printf "\t"B[i][j]} ; print "" }}' gcta_kinship.grm.id gcta_kinship.grm > gcta_kinship.grm.addname.matrix
1. 软件安装
mamba create -n GWAS
mamba activate GWAS
# plink
conda install bioconda::plink
# emmax
wget https://csg.sph.umich.edu//kang/emmax/download/emmax-intel-binary-20120210.tar.gz
tar xvf emmax-intel-binary-20120210.tar.gz
# tassel
mamba install bioconda::tassel
# rMVP
install.packages("rMVP")
# GAPIT
https://github.com/jiabowang/GAPIT
2. 格式转化
表现数据格式如下
RNAseq_ID GD10Y
X07 45.21
X08 36.63
X02 30.25
X09 35.16
X06 43.14
X10 36.30
X11 41.05
X12 34.07
X13 48.23
emmax要求输入的数据不能是vcf格式,因此先使用plink进行格式转换。
plink --vcf snps.f4.withid.75samples.RNAseqID.vcf --maf 0.05 --geno 0.1 --recode 12 transpose --output-missing-genotype 0 --out snp_filter --set-missing-var-ids @:# --allow-extra-chr --keep-allele-order
--vcf snps.f4.withid.75samples.RNAseqID.vcf: 指定输入的 VCF 文件名。VCF (Variant Call Format) 是存储基因变异信息的标准格式。
--maf 0.05: 按次要等位基因频率(Minor Allele Frequency, MAF)进行过滤。此参数会移除数据集中 MAF 低于 5% 的所有 SNP(单核苷酸多态性)。
--geno 0.1: 按基因型缺失率进行过滤。此参数会移除在超过 10% 的样本中未能成功进行基因分型的 SNP。
--recode 12 transpose: 指定输出文件的格式。
'recode 12': 将基因型数据重新编码为数字:0 代表纯合参考基因型(如 A/A),1 代表杂合基因型(如 A/G),2 代表纯合备选基因型(如 G/G)。
'transpose': 将输出的矩阵转置,使得行为样本(Sample),列为 SNP。这在很多统计分析(如 R 语言)中是常见的输入格式。
--output-missing-genotype 0: 指定在输出文件中如何表示缺失的基因型。这里用 '0' 来代替缺失值。注意:这可能会与代表纯合参考基因型的 '0' 产生混淆,在后续分析中需要特别留意。
--out snp_filter: 指定输出文件的前缀。所有生成的文件都将以 "snp_filter" 开头,例如 snp_filter.ped。
--set-missing-var-ids @:#: 为那些在 VCF 文件中没有 ID(通常表示为 ".")的变异位点创建一个新的 ID。这个模板会使用染色体名(由@代表)和物理位置(由#代表)来生成 ID,格式如 chr1:12345。
--allow-extra-chr: 允许 VCF 文件中包含非标准或非常规的染色体名称(例如,scaffolds 或 contigs),这在处理非模式生物的基因组数据时非常有用。
--keep-allele-order: 强制 PLINK 保持 VCF 文件中定义的参考等位基因(REF)和备选等位基因(ALT)的顺序。默认情况下,PLINK 可能会为了方便内部处理而交换它们。
#snp_filter.log,snp_filter.nosex,snp_filter.tfam,snp_filter.tped。这四个文件是输出文件,后面会用到。
emmax要求性状特征的文本文件的顺序要和VCF文件的一致,这里用一个脚本来进行排序。
perl ort_pheno.pl snp_filter.tfam GD10_Triat.txt > Sort_GD10_Triat.txt
还要生成亲缘关系矩阵,gwas分析的时候作为矫正
~/Software/emmax/emmax-kin-intel64 -v -d 10 -o ./pop.kinship snp_filter
# snp_filter 输入输入文件的前缀
# -o 结果文件
3. GWAS分析
分析之前要强调以下,不同的软件支持不同的模型,并且在分析的时候可能会加入亲缘关系矩阵,PCA矩阵或者Q矩阵作为群体结构矫正。
| 软件 | 运算速度 | 模型数量 | 结果展示 |
|---|---|---|---|
| EMMAX | 速度较快,适用于大规模数据 | MLM | 输出简单,仅有文本信息 |
| TASSEL | 对大数据集处理较慢 | GLM、MLM、CMLM | 输出简单,仅有文本信息 |
| GAPIT | 较慢,适用于中等规模数据 | GLM、FarmCPU、MLM、GLM、CMLM、MMLM、SUPER | 曼哈顿图和QQplot图 |
| rMVP | 速度较快,支持大数据并行计算 | GLM、MLM、FarmCPU | 曼哈顿图和QQplot图 |
3.1 emmax
emmax只需要输入亲缘关系矩阵即可,软件会自动计算群体结构,不如要额外的PCA或Q矩阵。
~/Software/emmax/emmax-intel64 -v -d 10 -t snp_filter -p Sort_GD10_Triat.txt -k pop.kinship -o emmax.out
-v: 启用详细输出模式(verbose)。程序在运行时会打印更详细的进度和诊断信息,便于监控和调试。
-d 10: 设置调试信息的输出频率。具体来说,程序每处理 10 个标记(markers/SNPs)就会输出一次调试信息,方便跟踪分析进度。
-t snp_filter: 指定基因型文件的基本名称(basename)。EMMAX 通常需要 `.tped` 和 `.tfam` 格式的文件。此参数告诉程序查找名为 `snp_filter.tped(包含基因型数据)和
snp_filter.tfam`(包含家系或个体信息)的文件。
-p Sort_GD10_Triat.txt: 指定表型数据文件。Sort_GD10_Triat.txt 文件包含了分析中每个个体的表型值。文件中的个体必须与基因型文件(.tfam)中的个体相对应。
-k pop.kinship: 指定亲缘关系矩阵文件。pop.kinship 文件是一个方差-协方差矩阵,用于模拟个体间的遗传相关性。这是 EMMAX
混合模型方法的核心,通过它来校正群体分层和隐性亲缘关系,从而减少假阳性结果。
-o emmax.out: 指定输出文件的前缀。分析结果将保存在以 emmax.out 开头的多个文件中。其中,最主要的结果文件通常是 emmax.out.ps,包含了每个 SNP 的关联分析 p 值等信息。
还需要对原始的GWAS结果进行格式转换,方便后续绘制曼哈顿和qq图。
awk '{print $1"\t"$2"\t"$3"\t"$4"\t"}' snp_filter.tped |paste - emmax.out.ps | awk 'BEGIN{print "SNP\tCHR\tBP\tP"}{if($2==$5){print $2"\t"$1"\t"$4"\t"$NF}}' > emmax.out.ps.manht_input
3.2 tassle
tassle可以指定群体结构文件,即PCA或者Q矩阵,亲缘关系矩阵不需要。可以使用gml,mlm和cmlm模型GWAS分析。
# Triat_format
head ../GD10_Triat.txt
<Trait> C16_0
RNAseq_ID GD10Y
X07 45.21
X08 36.63
X02 30.25
X09 35.16
X06 43.14
X10 36.30
X11 41.05
X12 34.07
# 使用Q需要去除最后一列
awk '{print $1}' ../data/demo.nosex | paste - ../data/demo.3.Q |awk 'NR==1{printf "<Covariate>\n<Trait>";for(i=1; i<NF-1; i++){printf "\tQ"i}; printf "\n"}; {NF--; print $0}' > Qdata.txt
# Q format
head ../Qdata.txt
<Covariate>
<Trait> Q1 Q2
X01 0.078568 0.844972
X02 0.999980 0.000010
X03 0.714090 0.285900
X04 0.975353 0.000010
X05 0.000010 0.999980
X06 0.999980 0.000010
X07 0.000010 0.866252
X08 0.083799 0.860377
# 也可使用前3个主成分
sed 's/\s\+/\t/g' ../data/demo.PCA.eigenvec| cut -f 2-5 |awk 'NR==1{printf "<Covariate>\n<Trait>";for(i=1; i<NF; i++){printf "\tP"i}; printf "\n"}; {print $0}' > Qdata.txt
## 计算亲缘关系
run_pipeline.pl -Xms512m -Xmx10g -importGuess demo.sort.vcf -KinshipPlugin -method Centered_IBS -endPlugin -export kinship.txt -exportType SqrMatrix
# Run GWAS with gml model
run_pipeline.pl -Xms512m -Xmx10g \
-fork1 -vcf demo.sort.vcf \
-fork2 -t trait.txt \
-fork3 -q Qdata.txt \
-combine4 -input1 -input2 -input3 -intersect \
-FixedEffectLMPlugin -endPlugin \
-export glm_output
# Run GWAS with mlm model
run_pipeline.pl -Xms512m -Xmx20g \
-fork1 -vcf demo.sort.vcf \
-fork2 -t trait.txt \
-fork3 -q Qdata.txt \
-fork4 -k kinship.txt \
-combine5 -input1 -input2 -input3 -intersect \
-combine6 -input5 -input4 -mlm \
-mlmVarCompEst P3D -mlmCompressionLevel None\
-export mlm_output
# Run GWAS with cmlm model
run_pipeline.pl -Xms512m -Xmx20g \
-fork1 -vcf demo.sort.vcf \
-fork2 -t trait.txt \
-fork3 -q Qdata.txt \
-fork4 -k kinship.txt \
-combine5 -input1 -input2 -input3 -intersect \
-combine6 -input5 -input4 -mlm \
-mlmVarCompEst P3D -mlmCompressionLevel Optimum \
-export cmlm_output
# 调整格式,方便绘图
awk '{print $2"\t"$3"\t"$4"\t"$7}' cmlm_output2.txt | awk '$NF ~ /[0-9]+/ || NR == 1 ' > cmlm_output.manht_input
3.3 GAPIT
GPAIT给我的第一印象就是一个字“全”,是所有GWAS分析中支持的模型最多的,缺点就是速度较慢。并且不需要要输入亲缘关系矩阵和群体结构文件,软件会自动计算并进行矫正。输入的基因型数据要求是hmp格式的。此外,该软件是个R包。
# Trait dir format
head trait.txt
Taxa GD10 GD5 ZG
X01 35.22 39.1 197.0
X02 30.25 32.53 224.0
X03 39.27 49.52 274.3
X04 NA 57.6 265.5
X05 40.95 42.8 185.0
X06 43.14 45.74 238.5
X07 45.21 46.22 184.5
X08 36.63 37.66 188.5
X09 35.16 36.08 203.0
## 准备基因型数据
run_pipeline.pl -Xms1G -Xmx5G -importGuess ../data/demo.vcf -ExportPlugin -format HapmapDiploid -saveAs demo.hmp.txt
sed -i '1s/#//g' demo.hmp.txt
运行GAPIT的R脚本
library(GAPIT)
## 读取表现数据
myY=read.table(file="pheno.txt", head = TRUE)
## 读取基因型数据
myG=read.table(file="demo.hmp.txt", head = F)
## 运行gapit
myGAPIT <- GAPIT(
Y=myY[,c(1,2)], # 表型
G=myG, #
model=c("Blink","FarmCPU"), # MLM, GLM, CMLM, MMLM, SUPER, FarmCPU, Blink
kinship.algorithm = "VanRaden", # EMMA, Loiselle, VanRaden, Zhang
PCA.total=5,
file.output=T
)
#save(myGAPIT, file="./myGAPIT.Rdata")
3.4 rMVP
同样是一个做GWAS分析的R脚本,相比于GAPIT,模型较少,但是速度快,出图好看,直接就可以输入VCF文件。同样不刚需亲缘关系矩阵和群体结构文件,软件会自动结算。
# Trait dir format
head trait.txt
Taxa GD10 GD5 ZG
X01 35.22 39.1 197.0
X02 30.25 32.53 224.0
X03 39.27 49.52 274.3
X04 NA 57.6 265.5
X05 40.95 42.8 185.0
X06 43.14 45.74 238.5
X07 45.21 46.22 184.5
X08 36.63 37.66 188.5
X09 35.16 36.08 203.0
运行rMVP的R脚本
# Run GWAS with rMVP
library(rMVP)
# genotype数据存储为MVP格式
MVP.Data(fileVCF="./demo.vcf",
#fileHMP="hapmap.txt",
#fileBed="plink",
filePhe="trait.txt",
fileKin=F,
filePC=F,
out="mvp",
ncpus = 5
)
# 读取genotyp和表型数据
genotype <- attach.big.matrix("mvp.geno.desc")
map <- read.table("mvp.geno.map" , head = TRUE)
phenotype <- read.table("mvp.phe",head=TRUE)
## 可单独计算Kinship和PC
K <- MVP.K.VanRaden(genotype)
PC <- MVP.PCA(genotype,pcs.keep = 5)
write.table(K, file = "kinship.matrix", sep="\t",quote = F, row.names = F, col.names =
F)
write.table(PC, file = "PC.matrix", sep="\t",quote = F, row.names = F, col.names = F)
# 支持模型 GLM, MLM 和 FarmCPU.
imMVP <- MVP(
phe=phenotype,
geno=genotype,
map=map,
#K=Kinship,
#CV.GLM=Covariates, ##if you have additional covariates, please keep there open.
#CV.MLM=Covariates,
#CV.FarmCPU=Covariates,
nPC.GLM=3, ## GLM 模型PC数
nPC.MLM=3, ## MLM 模型PC数
nPC.FarmCPU=3, ## FarmCPU模型中加入的PC数量
ncpus=5,
vc.method="BRENT", ##only works for MLM,"EMMA" or "HE" or "BRENT"
maxLoop=10, ## FarmCPU 迭代次数
method.bin="static", ## "FaST-LMM", "static" (#only works for FarmCPU)
#permutation.threshold=TRUE,
#permutation.rep=100,
threshold=0.05, # 0.05/marker size
method=c("GLM", "MLM", "FarmCPU"),
file.output=T
)
#save(imMVP, file = "imMVP.Rdata")
4. 可视化
可视化选择CMplot,简单并且美观!
输入文件格式如下
# GWAS result dir format
head Plot_GD10.FarmCPU.txt
SNP CHROM POS GD10
SNP1 chr01 20168 0.322959574748127
SNP2 chr01 20214 0.673619517210625
SNP3 chr01 20233 0.177721400235444
SNP4 chr01 20240 0.0547135109062588
SNP5 chr01 20264 0.725029263883326
SNP6 chr01 20270 0.0547135109062588
SNP7 chr01 20337 0.0541283609666494
SNP8 chr01 20339 0.179058722450606
SNP9 chr01 20354 0.44060986323047
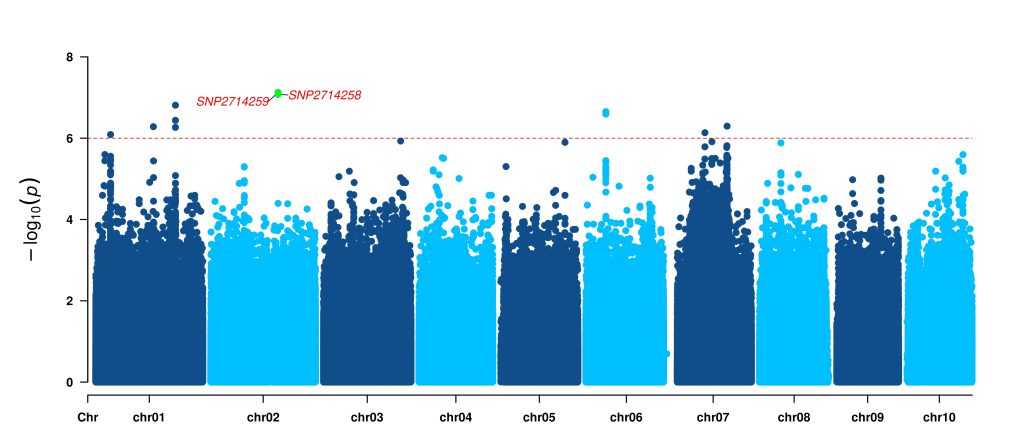
# Highlight gene list
head Plot_GD10.FarmCPU_highlight_gene.txt
SNP2714258
SNP2714259
绘图
library(CMplot)
# Exprot highlight gene list
GD10_gene_df <- read.table("Plot_GD10.FarmCPU_highlight_gene.txt")
GD10_gene_vector <- GD10_gene_df$V1
# Exprot GWAS result
GD10 <- read.table("Plot_GD10.FarmCPU.txt", header = TRUE)
# Manhattan Plot
CMplot(GD10, plot.type="m",LOG10=TRUE,col=c("dodgerblue4","deepskyblue"),highlight=GD10_gene_vector,
highlight.col=rep(c("green"),length=length(GD10_gene_vector)),highlight.cex=1, highlight.text=GD10_gene_vector,
highlight.text.col=rep("red",length(GD10_gene_vector)),threshold=1e-6,threshold.lty=2,
amplify=FALSE,file="jpg",dpi=300,verbose=TRUE,width=14,height=6,highlight.text.cex=1)
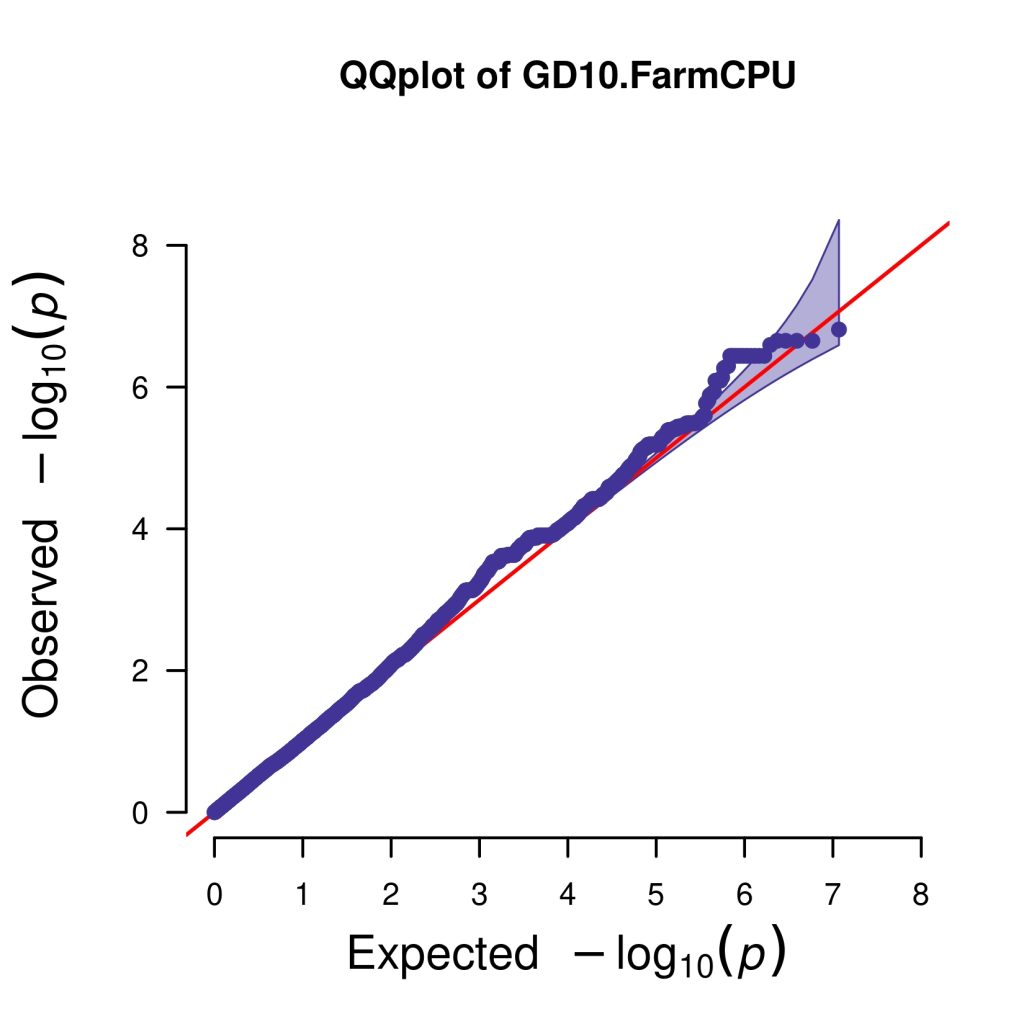
# QQplot
CMplot(GD10,plot.type="q",box=FALSE,file="jpg",file.name=NULL,dpi=300,
conf.int=TRUE,conf.int.col=NULL,threshold.col="red",threshold.lty=2,
file.output=TRUE,verbose=TRUE,width=5,height=5)