单倍型基因组挂载神器-Haphic
1. 软件官网
2. 软件下载
# 下载Haphic
git clone https://github.com/zengxiaofei/HapHiC.git
# 通过conda的yml文件安装依赖环境
conda env create -f HapHiC/conda_env/environment_py310.yml
# 启动Haphic环境
conda activate haphic # or: source /path/to/conda/bin/activate haphic
检查软件依赖
/path/to/HapHiC/haphic check
# 测试打印软件帮助信息
/path/to/HapHiC/haphic -h
3. 软件使用
将Hi-C测序数据比对到组装好的基因组上,以构建BAM文件
# 比对
bwa index asm.fa
bwa mem -5SP -t 28 asm.fa /path/to/read1_fq.gz /path/to/read2_fq.gz | samblaster | samtools view – -@ 14 -S -h -b -F 3340 -o HiC.bam
# 通过MAPQ >1 和NM < 3筛选比对结果
/path/to/HapHiC/utils/filter_bam HiC.bam 1 –nm 3 –threads 14 | samtools view – -b -@ 14 -o HiC.filtered.bam
运行Haphic pipline
/path/to/HapHiC/haphic pipeline asm.fa HiC.filtered.bam nchrs –RE “AAGCTT” –threads 8
# nchr 改成预估的染色体树木
# –RE 是酶切位点的类型
4. 输出结果
01.cluster/corrected_asm.fa:更正后的 FASTA 格式的程序集。仅在启用装配体校正时生成此文件。
04.build/scaffolds.agp:一个 SALSA 风格的 AGP 文件,包含有关 corrected_asm.fa 中所有序列的脚手架分配、排序和方向信息。如果有任何嵌合重叠群被 HapHiC 校正,损坏的重叠群将被分配新的 ID。(后面的构建挂载图和Juicebox可视化调整需要用到的结果)
04.build/scaffolds.raw.agp:一个 YaHS 风格的 AGP 文件,包含有关 asm.fa 中所有序列的脚手架分配、排序和方向信息。不会为断开的重叠群分配新的 ID,但它们在原始重叠群中的起始和结束坐标将显示在第七列和第八列中。
04.build/scaffolds.fa:FASTA 格式的最终脚手架。(染色体挂载后的scaffold水平的基因组)
04.build/juicebox.sh:用于 Juicebox 可视化和管理的 shell 脚本。(快速生成Juicebox的输入文件脚本)
5. 后续补充
5.1 可视化挂载结果
# 原始的挂载结果
/path/to/HapHiC/haphic plot scaffolds.raw.agp HiC.filtered.bam
# 通过Juicebox调整后的挂在结果
/path/to/HapHiC/haphic plot out_JBAT.FINAL.agp HiC.filtered.bam
# 首次运行后,会生成一个contact_matrix.pkl,该文件可以替换为.agp文件,让plot运行的运行速度很快。
/path/to/HapHiC/haphic plot contact_matrix.pkl HiC.filtered.bam
# 还有几个可以让结果美观的参数
# –bin_size 1000 设置为只有大于1000bp的scaffold才能显示在图上,一般经过Juicebox调整后,会有一些并不是染色体,长度很小的scaffold,可以通过这个参数来让这些scaffold在图上不显示。
# –manual_vmax 热图颜色的范围
5.2 Juicebox 手动调整
# 生成 .hic和.assembly文件
bash juicebox.sh
# 根据调整后的.assembly文件重新生成.agp文件
/path/to/HapHiC/utils/juicer post -o out_JBAT out_JBAT.review.assembly out_JBAT.liftover.agp asm.fa
输出结果
out_JBAT.FINAL.agp:调整后的agp文件
5.3 使用参考基因组对Scaffold进行排序和定向
在制作HIC热图的时候,一般情况下都会把单倍型的同源染色体放在一起展示,这样看起来比较直接。Haphic可以将组装好的基因组通过另外一个近缘物种的参考基因组(染色体水平)进行比对,从而实现排序和定向。
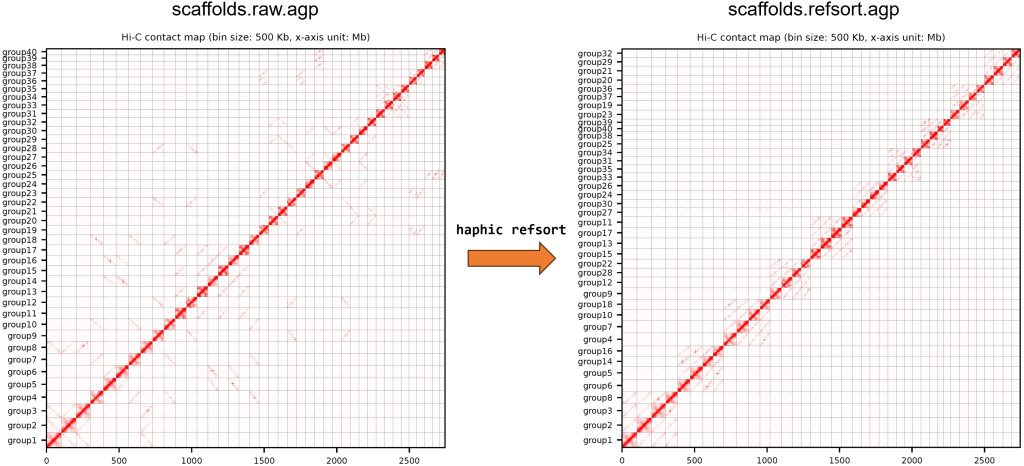
# 比对到参考基因组
minimap2 -x asm20 ref.fa asm.fa –secondary=no -t 28 -o asm_to_ref.paf
# 生成新的.agp文件(排序和定向后)
haphic refsort 04.build/scaffolds.raw.agp asm_to_ref.paf > scaffolds.refsort.agp