引言
在基因组注释中,真正复杂的往往不是单个预测软件的运行,而是如何将转录组、同源蛋白和从头预测等多种证据整合为可靠的基因模型。传统流程中,EVM 常被用于多证据加权整合,但其上下游步骤较多,证据预处理、权重设置和结果衔接也相对繁琐,尤其对新手并不友好。
随着高质量基因组不断增多,研究者对注释流程的要求也不再停留于“能跑通”,而是更加关注流程的完整性、自动化程度和结果稳定性。在这样的背景下,能够标准化整合多类证据、简化操作步骤的一站式注释工具,正变得越来越重要。
EviAnno 正是面向这一需求开发的工具。它聚焦于基因注释中最关键的证据整合环节,能够在保留多源证据优势的同时,减少繁琐的手动处理过程,提高注释效率。接下来,本文将结合实际使用流程,介绍 EviAnno 的安装、运行方法及结果解析。
Evianno
Evianno主要是基于rna-seq和同源蛋白的数据进行预测,但是也支持提供额外的gff文件证据,因此也可以结合de novo的结果。
安装
mamba install bioconda::eviann
准备输入文件
准备一个输入rna-seq数据的配置文件,可以是原始的fq和fa文件或者比对好的bam文件
/path/rna1_R1.fastq /path/rna1_R2.fastq /path/IsoSeq_rna.fastq
/path/rna1_R1.fastq /path/rna1_R2.fastq
/path/rna2_R1.fa /path/rna2_R2.fa fasta
/path/rna3.bam bam
合并uniprot数据库和近缘物种的蛋白文件作为同源蛋白
cat ChineseLong_pep_v3.fa Cucumber_64.pep Cucumber.CLv4.pep.fa Cucumber_gy14.pep Cucumber_w4.pep ~/sda1/database/Uniprot/uniprot_sprot.fasta > merge_pep.fasta
Helixer进行De novo注释
apptainer run --env PYTHONNOUSERSITE=1 --nv ~/sda1/database/helixer-docker_latest.sif Helixer.py --fasta-path Chrysanthemum_makinoi.fasta --lineage land_plant --gff-output-path Chrysanthemum_makinoi.gff --batch-size 60
注释
eviann.sh -t 48 -g quarTeT.genome.filled.fasta -r evianno.conf -p merge_pep.fasta -c Helixer.gff
# -g 基因组
# -t cpu数量
# -r rna-seq数据的配置文件
# -p 同源蛋白文件
# -c 额外的gff文件证据
quarTeT.genome.filled.fasta.pseudo_label.gff:是最后注释的gff文件
quarTeT.genome.filled.fasta.proteins.fasta:蛋白文件,存在可变剪切
quarTeT.genome.filled.fasta.transcripts.fasta:转录本文件
和EVM流程的注释结果比较
EVM流程注释到了23542个基因,Evianno注释到了28655个基因
EVM流程注释花费19个小时,Evianno花费4个小时
EVM辗转N多软件,Evianno一条命令完成
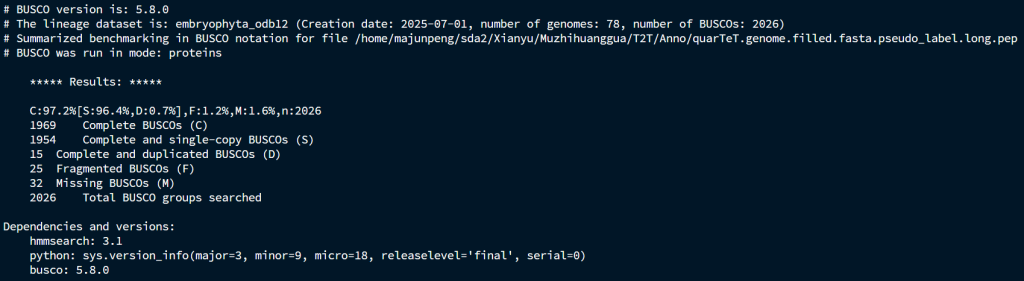
Evianno流程的BUSCO
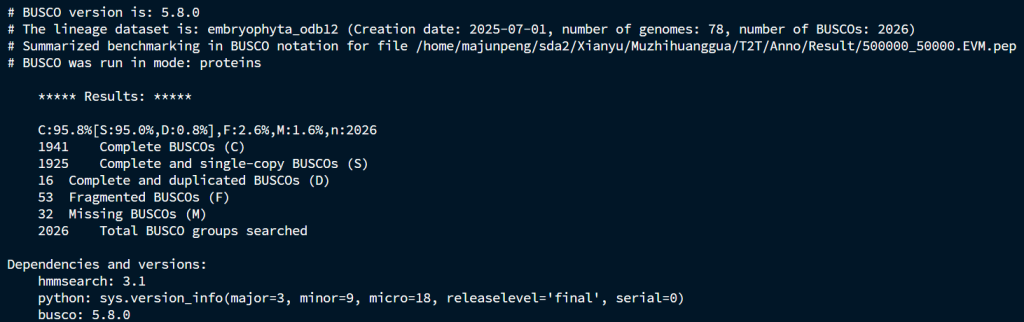
EVM流程的BUSCO
结语
无论是从运行的时间、操作难度和注释结果,Evianno都完全碾压EVM流程,以后可以直接换流程了。
欢迎加入交流群一起学习,添加好友和申请加入群时,请备注*方向+名称+单位”,谢谢!

