引言
传统系统发育分析通常以分叉式系统发育树为框架,假设物种演化仅通过谱系分化完成。然而,在真实的进化过程中,杂交(hybridization)、基因渗入(introgression)、水平基因转移以及不完全谱系分选(ILS)等过程普遍存在,尤其在植物多倍化类群和快速辐射分化的谱系中更为常见。这些过程会导致不同基因树之间出现显著冲突,使单一物种树难以完整刻画真实的演化历史。
为了解析这种复杂的“网状进化”模式,PhyloNetworks 提供了基于多基因树信息推断系统发育网络的统计方法。其核心算法 SNaQ 在共祖模型框架下,结合伪似然估计,在考虑 ILS 的同时识别潜在的杂交节点及其贡献比例,从而在传统物种树之外构建包含杂交事件的系统发育网络。因此,PhyloNetworks 为解析多倍体起源、近缘物种基因流及复杂演化历史提供了重要工具,使系统发育分析从“树”拓展至“网”。
PhyloNetworks
安装
PhyloNetworks是基于julia制作的,因此在下载PhyloNetworks之前,要先下载并配置julia
curl -fsSL https://install.julialang.org | sh
# 进入julia
julia
进入julia后,下载PhyloNetworks及其依赖软件
using Pkg # to use functions that manage packages
Pkg.add("PhyloNetworks") # to download & install package PhyloNetworks
Pkg.add("PhyloPlots")
Pkg.add("RCall") # packaage to call R from within julia
由于PhyloNetworks最新版本的一些命令发生了变化,所以这里指定下载0.16.4版本
Pkg.add(Pkg.PackageSpec(;name="PhyloNetworks", version="0.16.4"))
准备输入文件
PhyloNetworks进行网状进化分析的输入文件有两个(1)基因树(2)物种树(串联法或并联法都可以),需要注意的是,基因树和物种树都是不需要定根的。
网状进化分析
这里我们通过一个julia脚本,“raxmljulia.jl”进行网状进化分析。这里一共运行5次,只有第一次使用的是我们提供得物种树,后面的每一次都是基于上一次得结果。
using PhyloNetworks
using PhyloPlots
#raxmlCF = readTree2CF("Gene_tree.tre", writeTab=false, writeSummary=false)
trees = readMultiTopology("Prune_Gene_tree.tre")
q,t = countquartetsintrees(trees)
df = writeTableCF(q,t)
raxmlCF = readTableCF(df)
astraltree = readTopology("Species.tre")
using Distributed
addprocs(30)
@everywhere using PhyloNetworks
net0 = snaq!(astraltree, raxmlCF, hmax=0, filename="net0_raxml", seed=123, runs=50)
net1 = snaq!(net0, raxmlCF, hmax=1, filename="net1_raxml", seed=456, runs=50)
net2 = snaq!(net1, raxmlCF, hmax=2, filename="net2_raxml", seed=789, runs=50)
net3 = snaq!(net2, raxmlCF, hmax=3, filename="net3_raxml", seed=456, runs=50)
net4 = snaq!(net3, raxmlCF, hmax=4, filename="net4_raxml", seed=789, runs=50)
# Prune_Gene_tree.tre gene tres
# Species.tre species tree
运行脚本,进行网状进化分析
julia raxmljulia.jl
绘制网状进化图
先置根
using PhyloNetworks
using PhyloPlots
using RCall
net0 = readSnaqNetwork("net0_raxml.out")
rootatnode!(net0, "Campanula_reverchonoii_no_CAM121")
net1 = readSnaqNetwork("net1_raxml.out")
rootatnode!(net1, "Campanula_reverchonoii_no_CAM121")
net2 = readSnaqNetwork("net2_raxml.out")
rootatnode!(net2, "Campanula_reverchonoii_no_CAM121")
net3 = readSnaqNetwork("net3_raxml.out")
rootatnode!(net3, "Campanula_reverchonoii_no_CAM121")
net4 = readSnaqNetwork("net4_raxml.out")
rootatnode!(net4, "Campanula_reverchonoii_no_CAM121")
绘图
R"pdf"("plot4-net0123.pdf", width=28, height=12)
plot(net0, :R);
plot(net1, :R, showGamma=true, style=:majortree);
plot(net2, :R, showGamma=true, style=:majortree);
plot(net3, :R, showGamma=true, style=:majortree);
plot(net4, :R, showGamma=true, style=:majortree);
R"dev.off()";
结果为五张图,分别对应五次分析的结果。
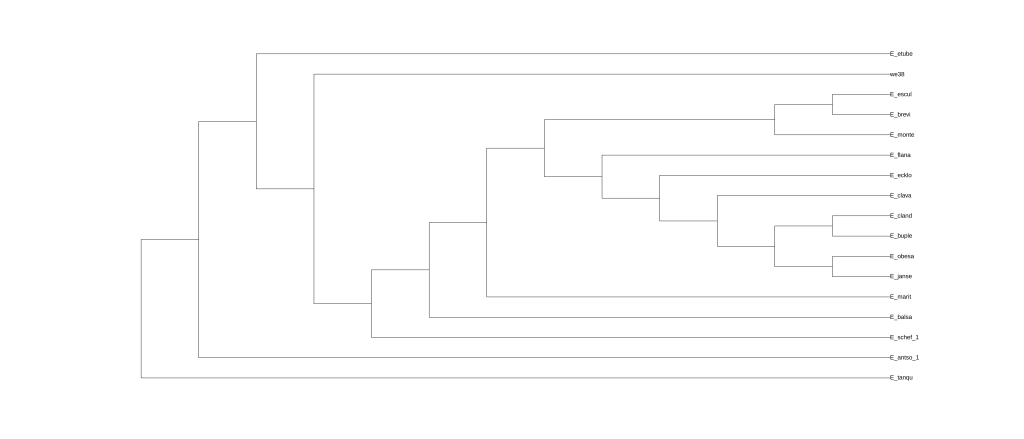
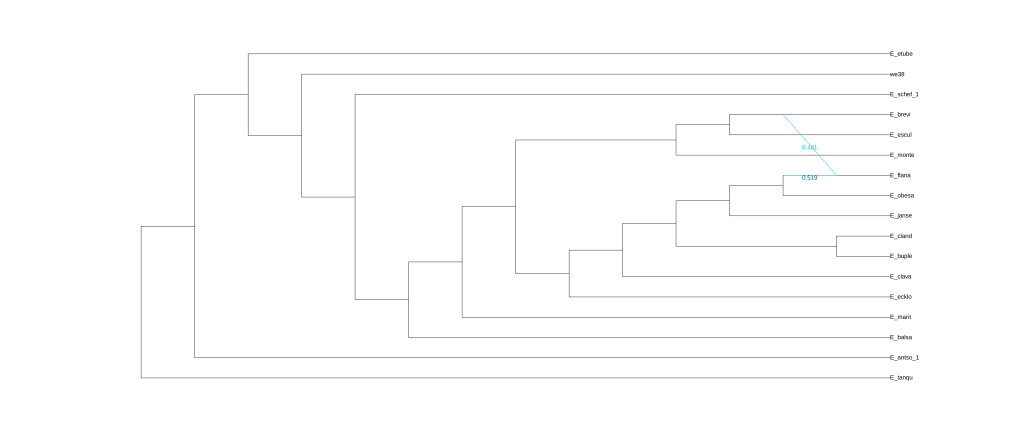
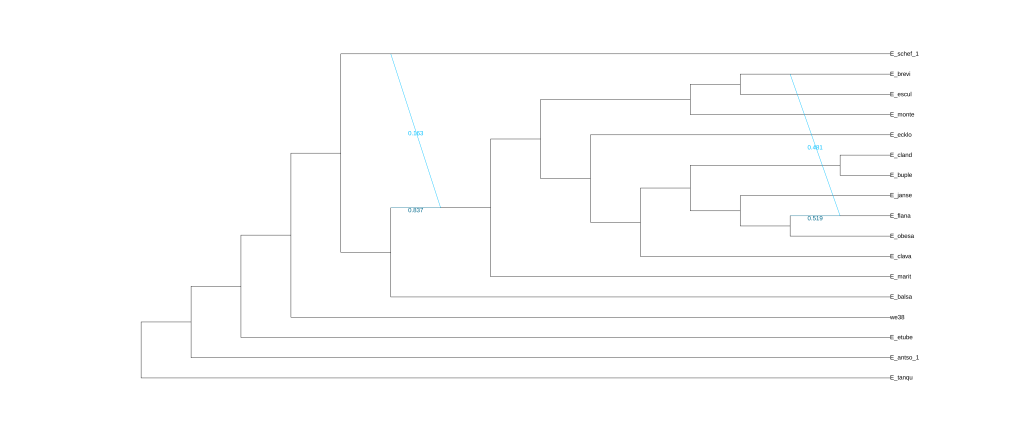
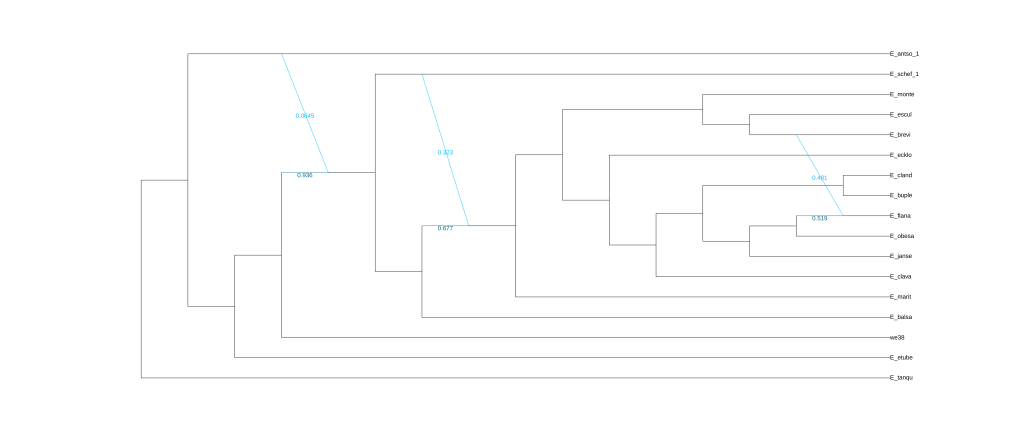
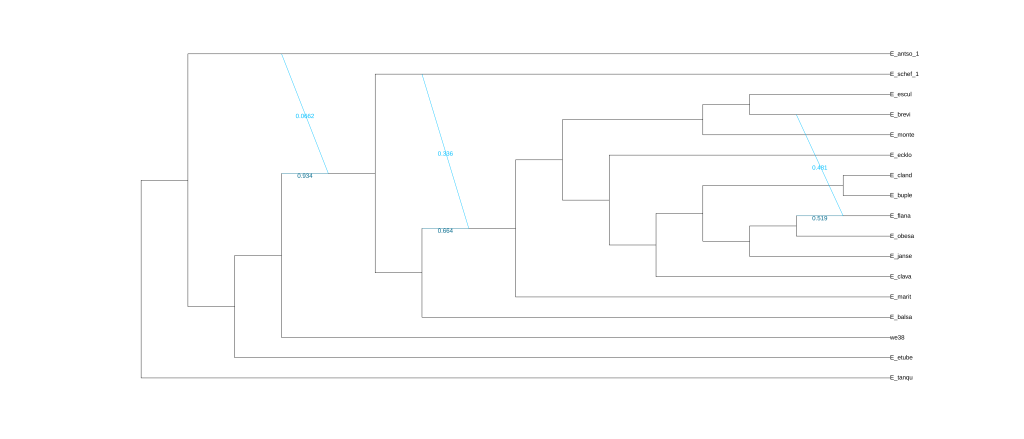
还可以使用Dendroscope进行可视化,将.out文件中Dendroscope后面的树复制粘贴到下框中。
评估最佳杂交次数
scores = [net0.loglik, net1.loglik, net2.loglik, net3.loglik, net4.loglik]
R"pdf"("score-vs-h0-4.pdf", width=5, height=5);
R"plot"(x=0:4, y=scores, type="o", xlab="number of hybridizations h", ylab="network score");
R"dev.off"();
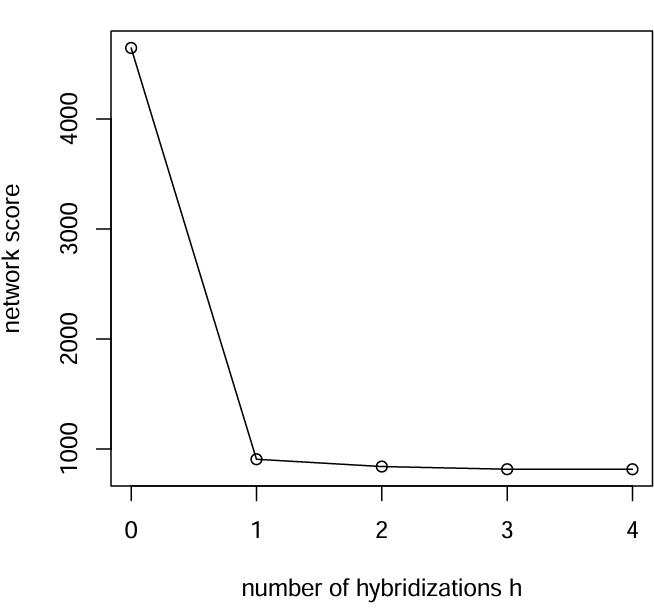
杂交次数从0-1变化很大,但是从1-2以及后续就没啥变化了,所以最佳的杂交次数就是1。