1. 前言
作为多倍体狂热爱好者的我,怎么能不学习亚基因组偏移分析呢,今天正好来复刻一下相关的分析和三元图。在这之前先说明一下三元图具体绘制标准。首先我们预先规定7个点(7种情况),
“Balanced”, 1/3, 1/3, 1/3,
“A_dominant”, 1, 0, 0,
“B_dominant”, 0, 1, 0,
“C_dominant”, 0, 0, 1,
“A_suppressed”, 0, 0.5, 0.5,
“B_suppressed”, 0.5, 0, 0.5,
“C_suppressed”, 0.5, 0.5, 0
随后根据三个基因组的相对表达量,分别计算该三元基因和这7个点的欧式距离,哪个距离最短,就被归为那种情况。
2. 分析
2.1 筛选三元基因
何为三元?其实就是每个亚基因组中1 vs 1 vs 1的基因,所以使用orthofinder筛选三个亚基因组的直系同源单拷贝基因即可。另外还要准备基因的表达量矩阵(这里是通过nf-core/rna-seq生成的)和三套亚基因组的gff文件。
orthofinder -f pep_dir -o Orthofinder_result -S diamond -a 10 -t 10
#生成三元基因列表
python get_OG_gene_list.py \
-i ./
-o OG_gene_list.txt
# 基于单拷贝基因结果,筛选三元基因,并且合并表达量矩阵
python generate_triad_expression_matrix.py \
--express Express.txt \
--og_list OG_gene_list.txt \
--gff_a Subgenome_A.gff \
--gff_b Subgenome_B.gff \
--gff_c Subgenome_B.gff \
--output Triad_Expression_Matrix.txt
# 按照三元基因的A+B+C=1进行标准化,生成相对表达量
python calculate_triad_ratios.py \
-i Triad_Expression_Matrix.txt
-o Triad_Ratios.txt
2.2 绘制三元图
library(tidyverse)
library(ggtern)
## ==========
## 1) 读取数据(按你上传文件路径)
## ==========
dat <- read.csv("Triad_Ratios.csv",
header = TRUE, check.names = FALSE, stringsAsFactors = FALSE)
## ==========
## 2) 统一列名(尽量不“猜”,而是自动识别)
## 你需要保证至少有:TriadID, Tissue,以及(ratio三列 或 expr三列)
## ==========
# 必需列:TriadID / Tissue(若你的列名大小写不同,请在这里改成你的真实列名)
# 例如如果是 "triad" 或 "OG" 之类,把下面改掉即可
stopifnot(all(c("TriadID", "Tissue") %in% colnames(dat)))
has_ratio <- all(c("A_ratio","B_ratio","C_ratio") %in% colnames(dat))
has_expr <- all(c("A_expr","B_expr","C_expr") %in% colnames(dat))
if (!has_ratio && !has_expr) {
stop("未找到比例列(A_ratio/B_ratio/C_ratio)或表达列(A_expr/B_expr/C_expr)。请检查表头。")
}
## 若没有 ratio,则用 expr 计算 ratio(加小伪计数避免 0/0)
if (!has_ratio) {
dat <- dat %>%
mutate(
A_ratio = (A_expr + 1e-6) / (A_expr + B_expr + C_expr + 3e-6),
B_ratio = (B_expr + 1e-6) / (A_expr + B_expr + C_expr + 3e-6),
C_ratio = (C_expr + 1e-6) / (A_expr + B_expr + C_expr + 3e-6)
)
}
## 如果存在总表达列(名字不固定),可选:用于过滤“表达的 triad”
# 文献:triad expressed if sum TPM > 0.5(按每个 tissue) :contentReference[oaicite:2]{index=2}
total_candidates <- c("Total", "Total_expr", "Sum", "TotalTPM", "TPM_sum", "triad_TPM")
total_col <- total_candidates[total_candidates %in% colnames(dat)][1]
if (!is.na(total_col)) {
dat <- dat %>% filter(.data[[total_col]] > 0.5)
} else {
# 若你确实想严格按文献过滤,但文件没有 total,可用 expr 现算一个:
if (has_expr) {
dat <- dat %>% mutate(Total_calc = A_expr + B_expr + C_expr) %>%
filter(Total_calc > 0.5)
}
}
## ==========
## 3) 文献的“理想类别点”(wheat32 经典定义)
## 六类:3 dominant + 3 suppressed;Balanced 单独作为中心点。
## 这是最常用、且与“顶点/边缘/中心”的几何含义一致。
## ==========
ideal <- tribble(
~category, ~A, ~B, ~C,
"Balanced", 1/3, 1/3, 1/3,
"A_dominant", 1, 0, 0,
"B_dominant", 0, 1, 0,
"C_dominant", 0, 0, 1,
"A_suppressed", 0, 0.5, 0.5,
"B_suppressed", 0.5, 0, 0.5,
"C_suppressed", 0.5, 0.5, 0
)
## ==========
## 4) 对每个 triad × tissue:计算到各理想点的欧氏距离,并取最短距离分类
## 这一步就是你引用的那段方法:rdist + shortest distance(逐组织) :contentReference[oaicite:3]{index=3}
## ==========
# 计算距离的函数(避免额外依赖 rdist 包)
euclid <- function(a,b,c, ia,ib,ic) sqrt((a-ia)^2 + (b-ib)^2 + (c-ic)^2)
plot_dat <- dat %>%
# 如果你文件里含有 "Average" 行,按文献 Fig.4d 通常用“每个组织”点,
# Average 不参与散点;若你想保留可注释下一行
filter(Tissue != "Average") %>%
rowwise() %>%
mutate(
# 计算到每个 ideal category 的距离
category = {
d <- ideal %>%
mutate(dist = euclid(A_ratio, B_ratio, C_ratio, A, B, C)) %>%
arrange(dist)
d$category[1]
}
) %>%
ungroup()
## ==========
## 5) 因子顺序(图例顺序与文献一致)
## ==========
plot_dat$category <- factor(
plot_dat$category,
levels = c("Balanced",
"A_dominant","B_dominant","C_dominant",
"A_suppressed","B_suppressed","C_suppressed")
)
## Tissue 的顺序:按你的真实组织名改(Root/Stem/Leaf/Bud 或 LB/LS/SH/RO/RH)
plot_dat$Tissue <- factor(plot_dat$Tissue)
## ==========
## 6) 配色/形状(你可按目标文献进一步微调)
## ==========
category_colors <- c(
"Balanced" = "grey70",
"A_dominant" = "#E41A1C",
"B_dominant" = "#377EB8",
"C_dominant" = "#4DAF4A",
"A_suppressed" = "#FB8072",
"B_suppressed" = "#80B1D3",
"C_suppressed" = "#B3DE69"
)
# 若你要严格复刻:Root/Stem/Leaf/Bud 可用 + ○ △ × 等
# 这里给一个通用映射;若你的组织名不同,按需要改 keys
tissue_shapes <- c(
"Root" = 3, # +
"Stem" = 1, # ○
"Leaf" = 2, # △
"Bud" = 4 # ×
)
## ==========
## 7) 绘制 ternary plot
## ==========
p <- ggtern(
data = plot_dat,
aes(x = A_ratio, y = B_ratio, z = C_ratio,
color = category, shape = Tissue)
) +
geom_point(size = 0.5, alpha = 0.7, stroke = 0.2) +
scale_color_manual(values = category_colors) +
scale_shape_manual(values = tissue_shapes, drop = FALSE) +
theme_bw() +
theme(
tern.panel.grid.major = element_line(color = "grey85"),
tern.panel.grid.minor = element_blank(),
legend.title = element_blank(),
legend.position = "right",
plot.margin = margin(10, 15, 10, 15, unit = "mm")
)+
labs(x = "Subgenome A", y = "Subgenome B", z = "Subgenome C")
## 计算百分比
triad_category <- plot_dat %>%
group_by(TriadID, category) %>%
summarise(n = n(), .groups = "drop") %>%
group_by(TriadID) %>%
slice_max(n, n = 1, with_ties = FALSE) %>%
ungroup()
category_pct_triad <- triad_category %>%
count(category) %>%
mutate(
percent = n / sum(n) * 100,
label = sprintf("%s (%.2f%%)", category, percent)
)
legend_labels <- setNames(
category_pct_triad$label,
category_pct_triad$category
)
p <- p +
guides(
color = guide_legend(
override.aes = list(
size = 3, # 图例中点的大小(推荐 2.5–3.5)
stroke = 0.3 # 图例中点的描边
)
),
shape = guide_legend(
override.aes = list(
size = 3,
stroke = 0.3
)
)
)
ggsave("Fig4d_Ternary_distance_based.pdf", p, width = 8, height = 4)
# 导出每个三元基因的分类
count_long <- plot_dat %>%
group_by(Tissue, category) %>%
summarise(n = n(), .groups = "drop")
count_wide <- count_long %>%
pivot_wider(
names_from = category,
values_from = n,
values_fill = 0
)
count_wide <- count_wide %>%
select(
Tissue,
Balanced,
A_dominant, B_dominant, C_dominant,
A_suppressed, B_suppressed, C_suppressed
)
write.csv(
count_wide,
file = "Triad_expression_bias_by_tissue_counts.csv",
row.names = FALSE
)
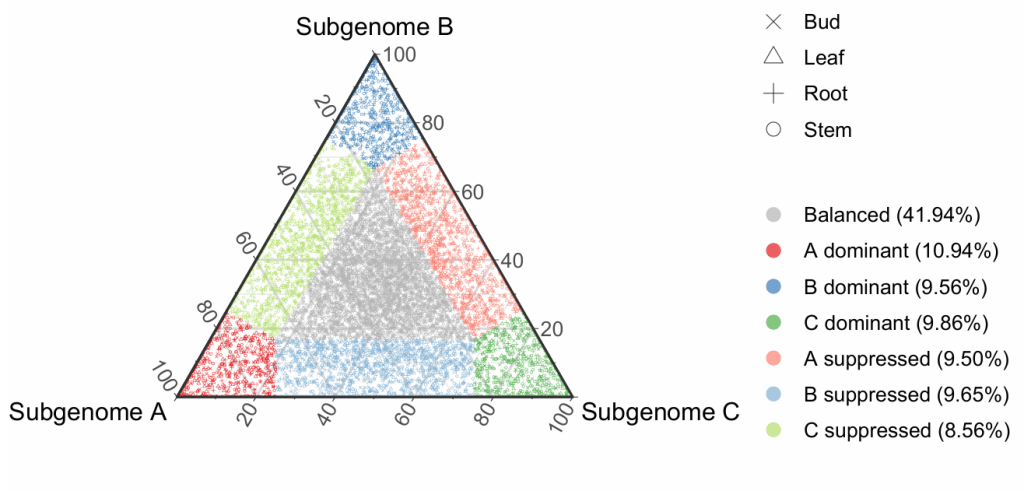
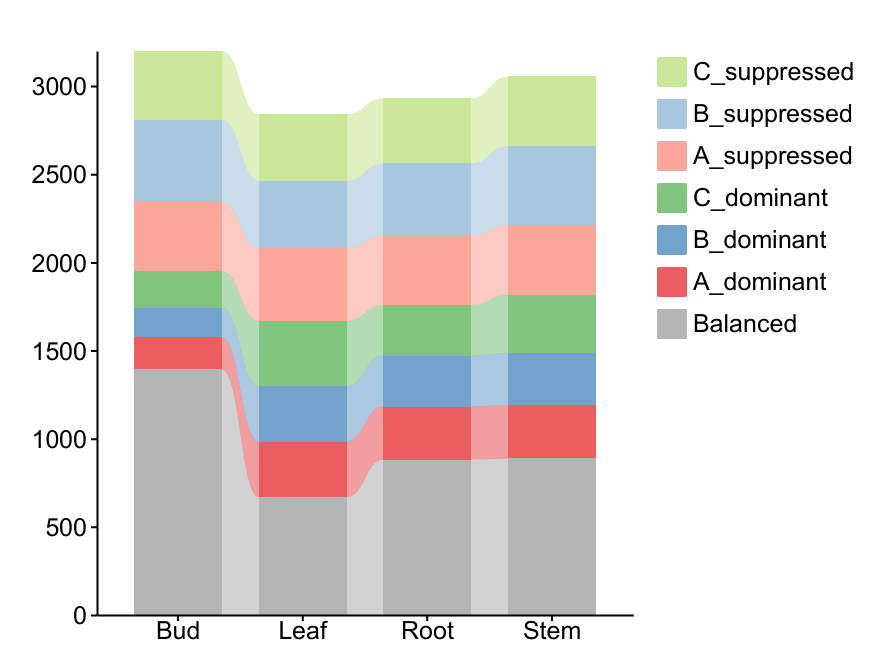
很奇怪,文献中对于Balanced的比例很高,有80%,但是我研究的这个物种很少,只有40%,暂时不知道是什么原因。另外,通过导出的表格还可以绘制堆叠柱状图,看一下不同组织的亚基因组偏移是否有明显区别。