引言
在基因组研究中,单核苷酸多态性(SNP)长期被视为遗传变异的核心指标。然而,基因组不仅仅是由一个个单点碱基组成,更包含大规模的结构变异(Structural Variants, SVs),如大片段缺失、插入、倒位和易位等。这类变异常常对基因功能、调控网络以及性状形成具有深远影响,但由于技术和分析方法的限制,往往被传统 SNP 分析忽略。
SyRI(Synteny and Rearrangement Identifier)是一款专门用于基因组结构变异检测的工具,它能够在全基因组尺度上识别序列重排、拷贝数变异和同源区间的差异。通过对两条或多条基因组进行比对,SyRI 不仅可以精确定位结构变异的位置,还能揭示变异类型和影响范围,为深入理解基因组演化和功能提供可靠数据。
如果你想突破 SNP 分析的局限,全面捕捉基因组中的“大动作”,SyRI 无疑是最值得掌握的工具之一。本篇文章将从 SyRI 的核心原理出发,结合实际操作示例,带你快速上手结构变异的全基因组分析。
SYRI
SYRI主要是通过基因组 VS 基因组的比对文件,鉴定SV,并且可以配合下游的Plotsr绘制结构变异图。
安装
mamba install bioconda::syri
mamba install bioconda::plotsr
全基因组比对
要使用SYRI进行SV检测,第一步进行基因全基因组比对。这里需要注意的是,比对的两个基因组的同源染色体ID必须是一致的,并且方向也要是相同的。举个例子,参考基因组A的1号染色体是Chr01,那么Query基因组的1号染色体也必须是Chr01。我自己写了一个脚本,通过比对产生的paf文件,进行同源染色体ID的替换和方向的调整。如果需要,加我微信我发你。
全基因组比对
mm2plus -t 20 -x asm20 -c -eqx -secondary=no 97103_genome_v2.5.fa XG0378_genome.fa > mm2plus.paf
基于比对的结果,替换Query基因组的同源染色体ID,并统一序列的方向
python3 ~/Script/syri/rename_chromosomes_by_paf_1.py -p mm2plus.paf -f XG0378_genome.fa -o Rename_XG0378_genome.fa
再次进行全基因组比对
mm2plus -ax asm5 -t 20 --eqx 97103_genome_v2.5_chr.genome.fasta Rename_XG0378_genome.fa > out.sam
SYRI鉴定SV
syri -c out.sam -r 97103_genome_v2.5_chr.genome.fasta -q Rename_XG0378_genome.fa -k -F S --nc 20 --prefix syri_
# -c 全基因组比对结果
# -r 参考基因组
# -q query基因组
# -k 保留过程文件
# -F 输入文件类型,这里指定为sam文件
# --nc cpu数量
# --prefix 输出文件前缀
最重要的输出文件是syri_syri.out文件,记录了每个SV的类型和位置信息。
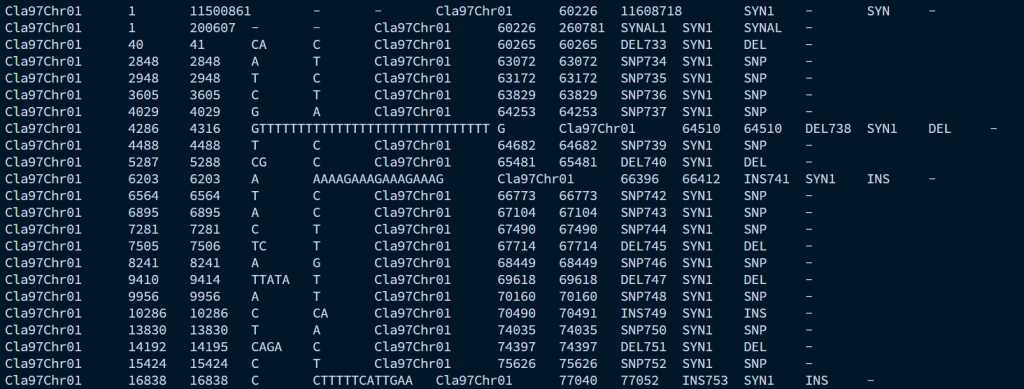
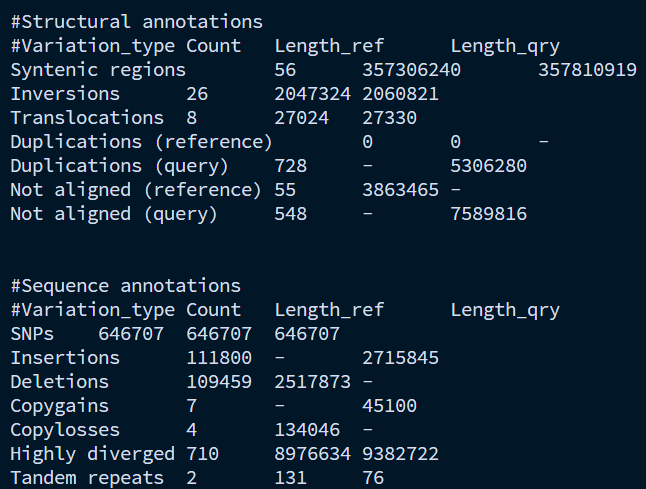
syri_syri.summary统计了每种SV的数量和长度
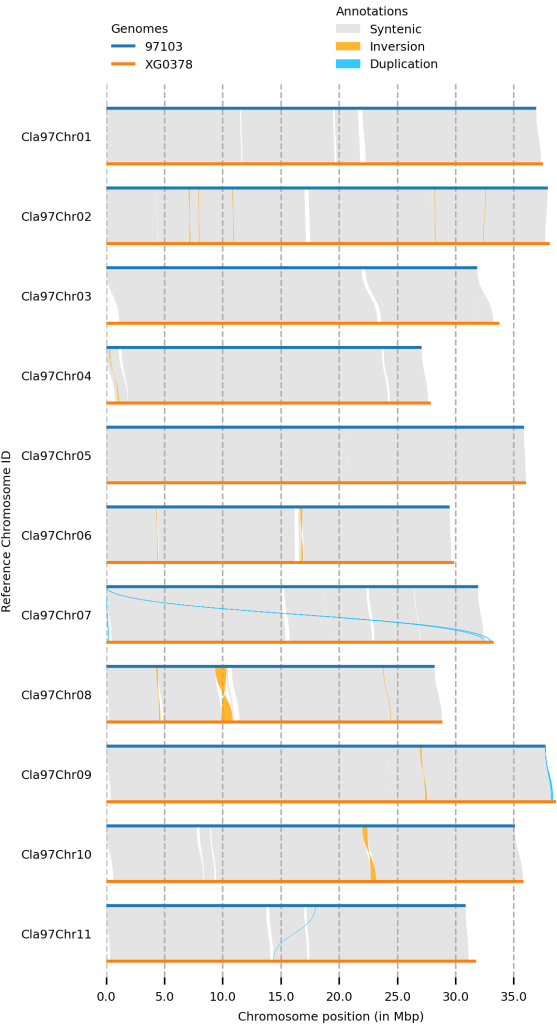
Plotsr
鉴定SV后,还可以绘制全基因组结构变异图,首先要先配置一个基因组信息文件,记录每个基因组的名称和文件信息。第一列为基因组文件名,第二列为基因组名称,第三列为一些配置信息。
97103_genome_v2.5_chr.genome.fasta 97103 lw:1.5
Rename_XG0378_genome.fa XG0378 lw:1.5
绘图
plotsr --sr syri_syri.out --genomes genome.txt -o output_plot.png -H 8 -W 4
# --sr syri的.out输出结果
# --genomes 基因组信息文件
# -o 输出结果图片
# -H 图片的高度
# -W 图片的宽度
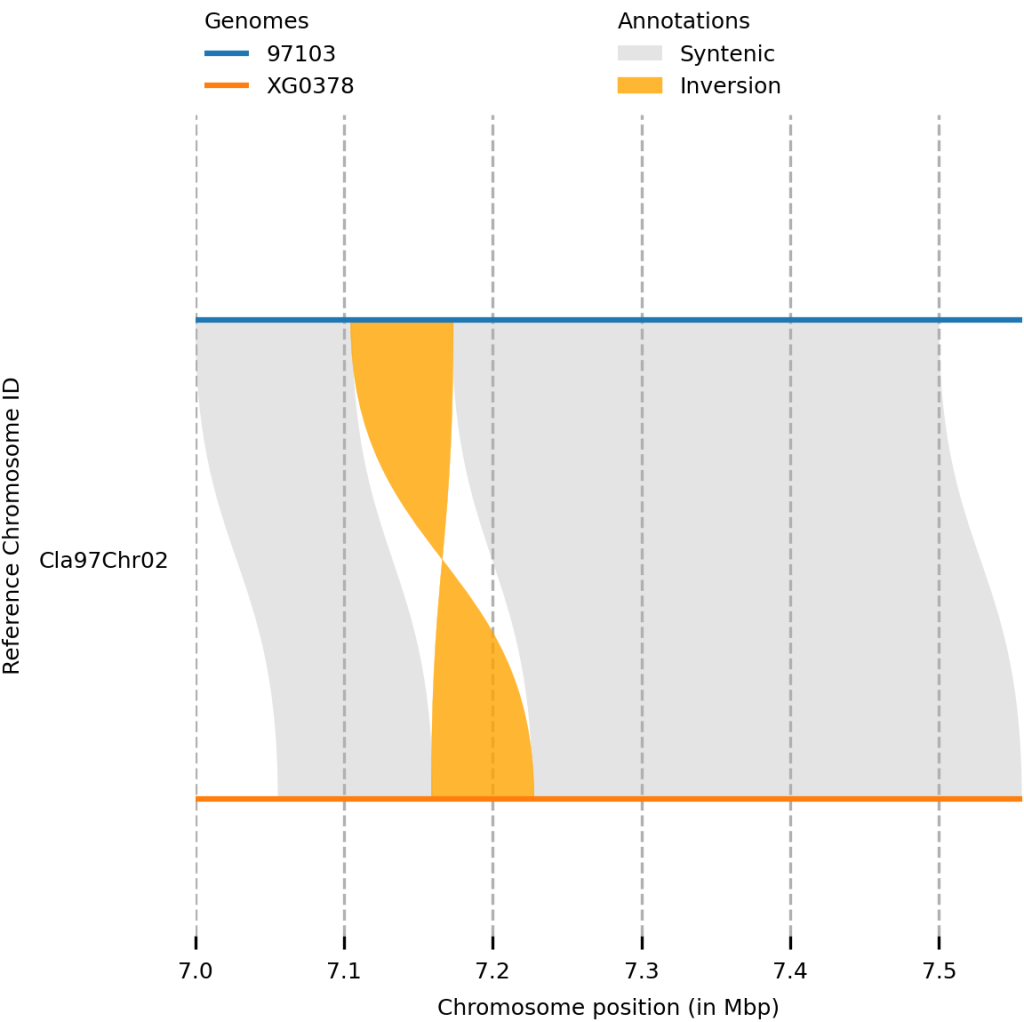
还可以绘制局部区域
plotsr --sr syri_syri.out --genomes genome.txt -o output_plot.png -H 4 -W 4 --reg 97103:Cla97Chr02:7000000-7500000
还可以在基因组的任何位置,加上各种标记,配置一个标记信息文件。
#chr start end genome_id tags
Cla97Chr02 7104444 7173464 97103 mt:v;mc:black;ms:3;tt:Inversion 1;tp:0.02;ts:8;tf:Arial;tc:black
plotsr --sr syri_syri.out --genomes genome.txt -o output_plot.png -H 4 -W 4 --reg 97103:Cla97Chr02:7000000-8000000 --markers markers.bed
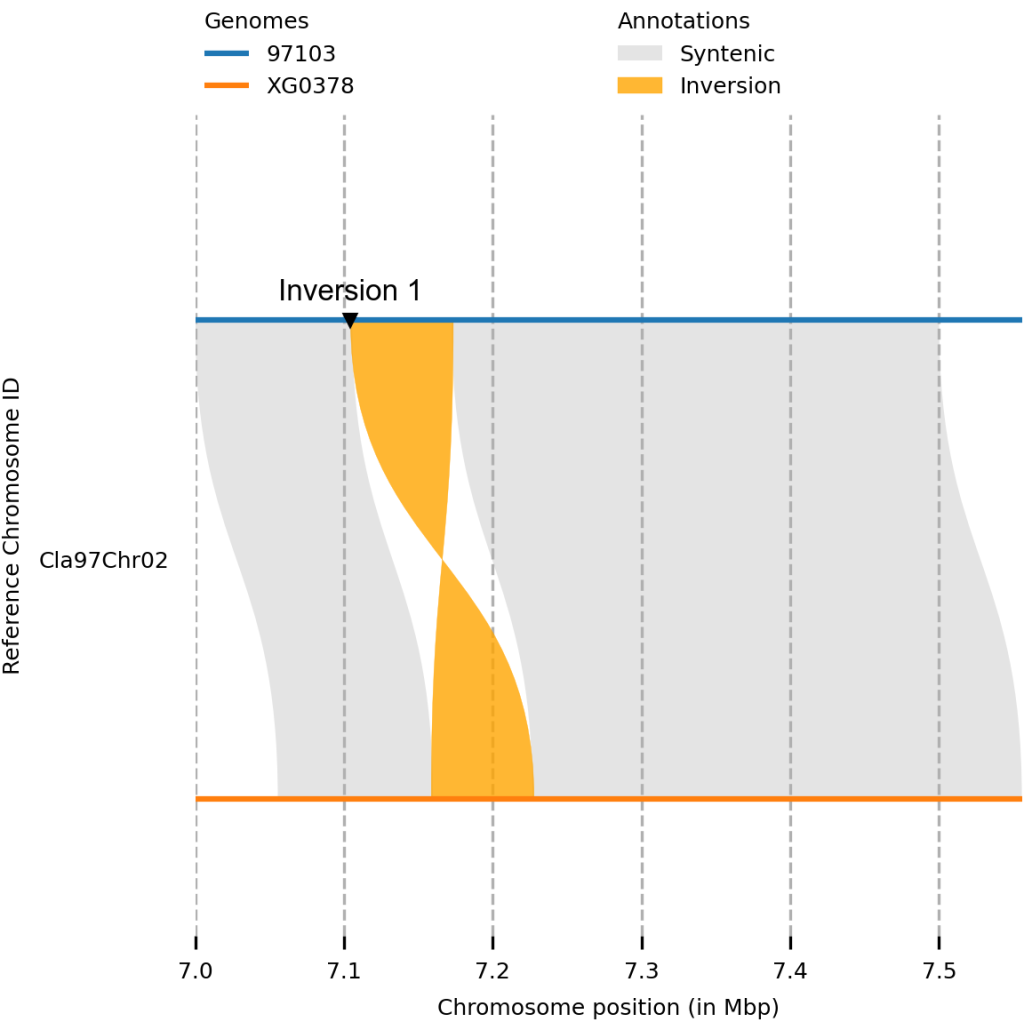
使用两种不同的标记,标记两个倒位。
#chr start end genome_id tags
Cla97Chr02 7104444 7173464 97103 mt:v;mc:black;ms:3;tt:Inversion 1;tp:0.02;ts:8;tf:Arial;tc:black
Cla97Chr02 7923535 7957505 97103 mt:o;mc:black;ms:3;tt:Inversion 2;tp:0.02;ts:8;tf:Arial;tc:red
plotsr --sr syri_syri.out --genomes genome.txt -o output_plot.png -H 4 -W 4 --reg 97103:Cla97Chr02:7000000-8000000 --markers markers.bed
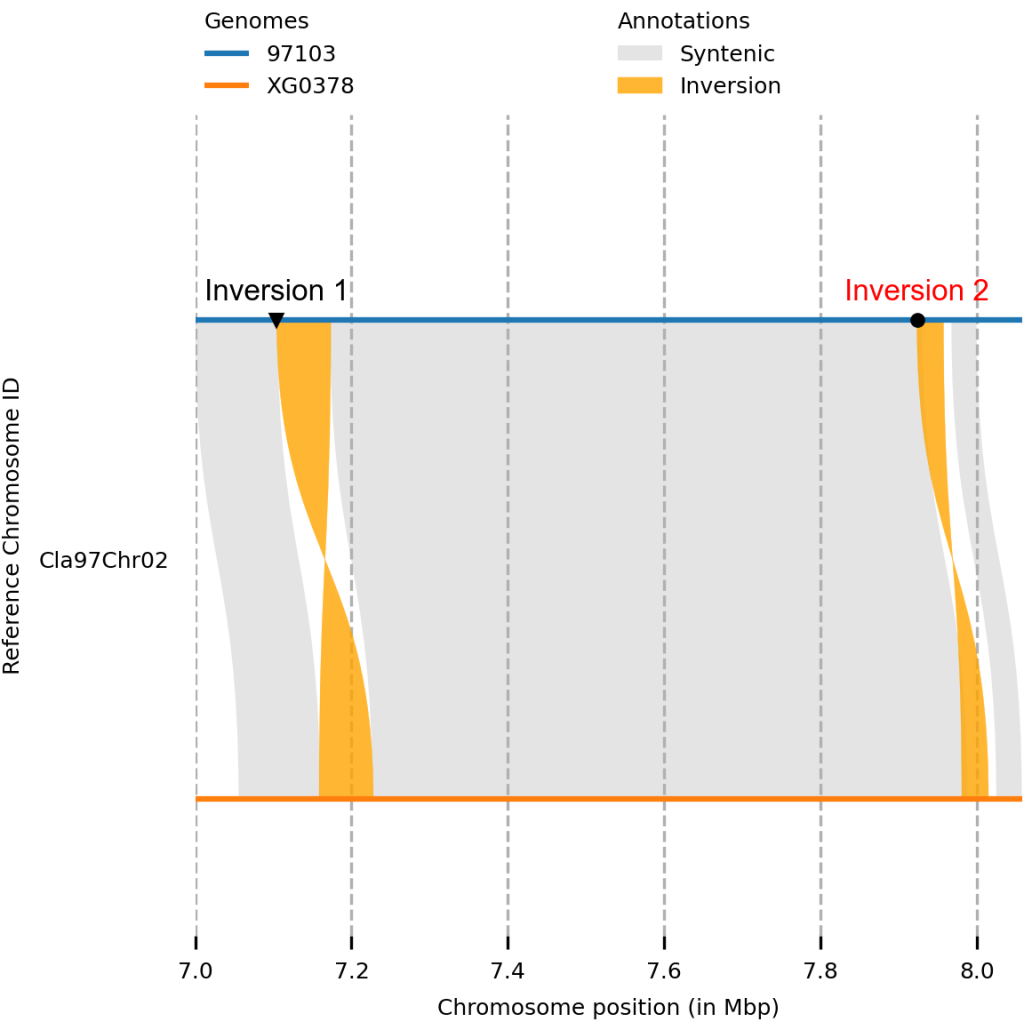
结语
SYRI+Plotsr组合其实我早就做过了,正好今天有一位老师想做一个SV分析,看一下两个基因组所有倒位类型的SV信息,借着这个机会介绍一下这个流程。
欢迎加入交流群一起学习,添加好友和申请加入群时,请备注“方向+名称+单位”,谢谢!

