引言
在开展比对、组装或变异检测等下游分析之前,二代测序数据的质量控制(QC)是不可省略的第一步。尽管 Illumina 等二代测序平台具有高通量和高性价比优势,但其测序过程仍会引入多种技术噪音,如 reads 3′ 端碱基质量下降、接头污染、PCR 扩增偏好及随机测序错误等。这些低质量序列若直接进入下游分析,将显著影响结果的可靠性:在参考基因组比对中会增加错配与多重比对,在变异检测中会引入假阳性信号,在 de novo 组装中则可能导致 contig 断裂或错误拼接。因此,质控的目的并非“美化数据”,而是最大限度降低技术误差,保留真实的生物学信息。基于此,FastQC、Fastp 和 Trimmomatic 等工具被广泛用于二代测序数据的质量评估与过滤,从不同层面构建起稳定、可靠的二代测序数据质控流程,为后续分析奠定坚实基础。
Fastqc
安装
mamba install bioconda::fastqc
质控
fastqc Clean_P636_YMiao_A01v1_S0-1_S1_L002_R1_001.fastq.gz \
Clean_P636_YMiao_A01v1_S0-1_S1_L002_R2_001.fastq.gz
每个测序文件会输出一个对应的html文件,记录了每个指标的检测情况。
结果解读
通过下列11项指标来进行测序质量的评估:
1. Basic Statistics
统计数据量和reads长度等基本指标
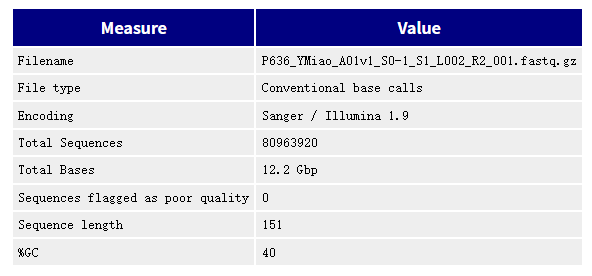
2. Per base sequence quality
用箱式图的方式展示数据质量,图中X轴每1个位置,都是该位置的所有序列的测序质量的统计。纵轴是质量得分,Q =-10*log10(p),p为测错的概率。所以一条reads某位置出错概率0.01时,其quality就是20。横轴是测序序列的位置。蓝色线是各个位置的平均值的连线。一般要求此图中,所有位置的10%分位数大于20,也就是常说的Q20过滤。。如果任何碱基质量低于10,或者是任何中位数低于25报警,如果任何碱基质量低于5,或者是任何中位数低于20报错。
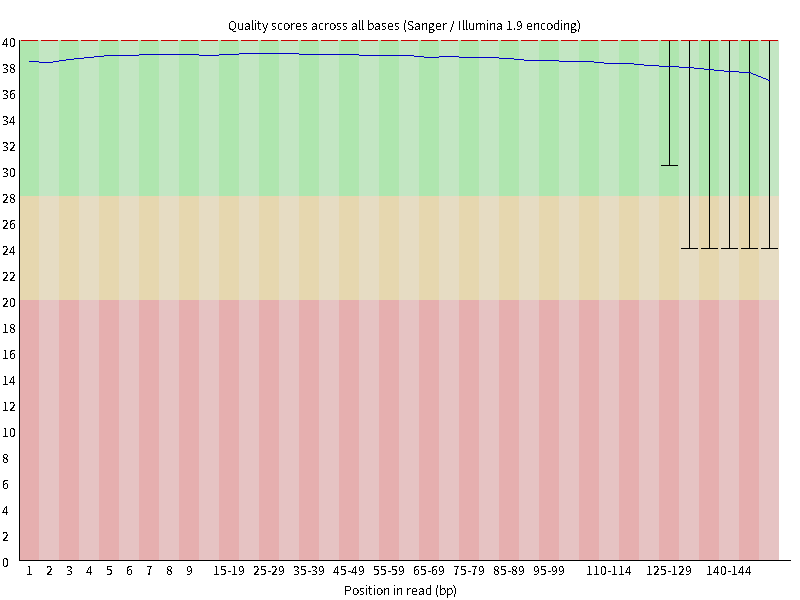
3. Per tile sequence quality
这一模块是检查在测序平台上,reads中每一个碱基位置在不同的测序小孔之间的偏离度,偏离度越高,碱基质量越差。纵轴表示测序小孔,蓝色表示低于平均偏离度,越红则说明偏离平均质量方差越多,也就是说质量越差,本图中都是蓝色表明质量很好。如果出现质量问题可能是短暂的,如有气泡产生,也可能是长期的,如在某一小孔中存在杂质。偏离度小于平均值2以上报警,偏离度小于平均值5以上不合格。
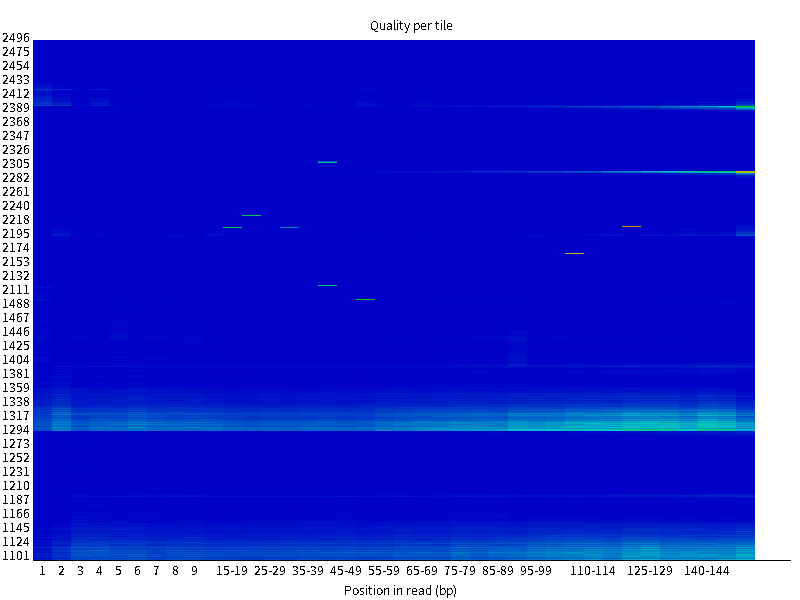
4. Per sequence quality scores
这是为了检测一部分质量特别差的reads,如果有则会在图上出现多个峰,如在测序仪边缘的reads。纵轴是reads数目,横轴是质量分数,代表不同Phred值对应了多少的reads。本图中,测序结果主要集中在高分中,证明测序质量良好。当峰值小于27(错误率0.2%)时警报,当峰值小于20(错误率1%)时不合格。
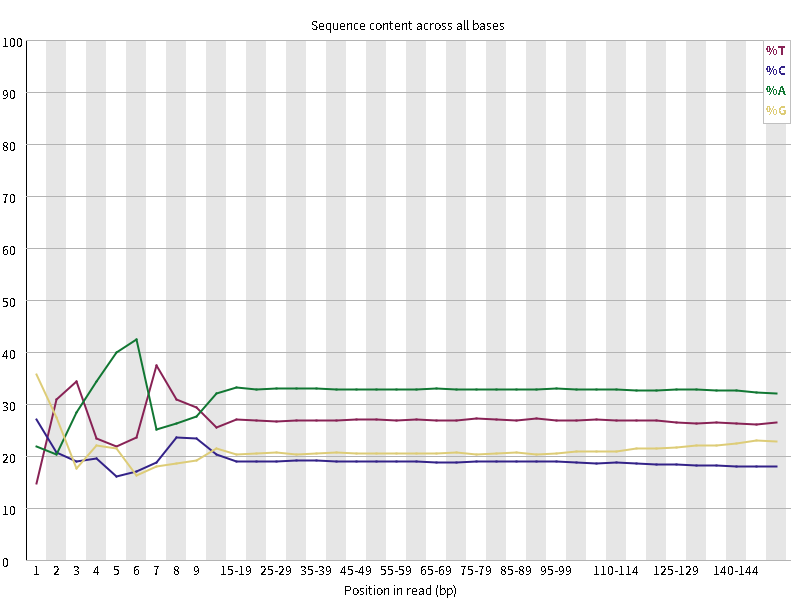
5. Per base sequence content
展示碱基含量分布,它根据碱基的位置对每个位置上的A,C,G,T的含量进行统计,横轴为位置,纵轴为百分比。正常情况下四种碱基的出现频率应该是接近的,而且没有位置差异。因此好的样本中四条线应该平行且接近。当部分位置碱基的比例出现bias时,即四条线在某些位置纷乱交织,往往提示我们有overrepresented sequence的污染。当所有位置的碱基比例一致的表现出bias时,即四条线平行但分开,往往代表文库有bias (建库过程或本身特点),或者是测序中的系统误差。当任一位置的A/T比例与G/C比例相差超过10%发出警报,超过20%则数据不合格。
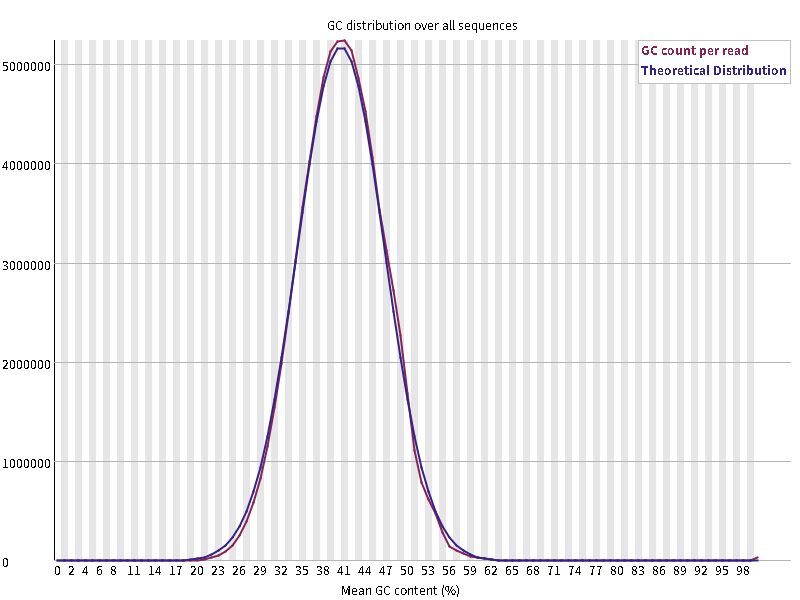
6. Per sequence GC content
红色曲线是实际的测序GC含量分布图,而蓝色曲线则是理论分布(正态分布,不过均值不一定都是50%,而是由平均GC含量推断的)。如果红色曲线形状存在比较大的偏差,往往是由于文库污染造成的。红色曲线越平滑越好,越接近蓝色曲线越好。形状接近正态但偏离理论分布的情况提示我们可能有系统偏差。偏离理论分布的reads超过15%时发出警报,超过30%时报不合格。
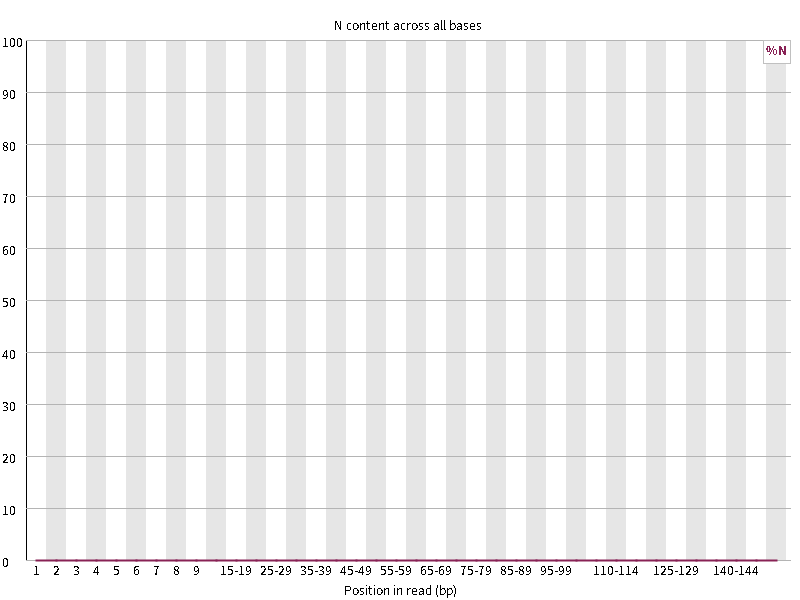
7. Per base N content
纵轴是百分含量,横轴是read的位置,当测序仪不能确切地测定出某一个碱基时就会标注为N,正常情况下N的比例是很小的,所以图上常常看到一条直线。当看到有峰时,说明测序出了问题。当任意位置的N的比例超过5%警报超过20%不合格。
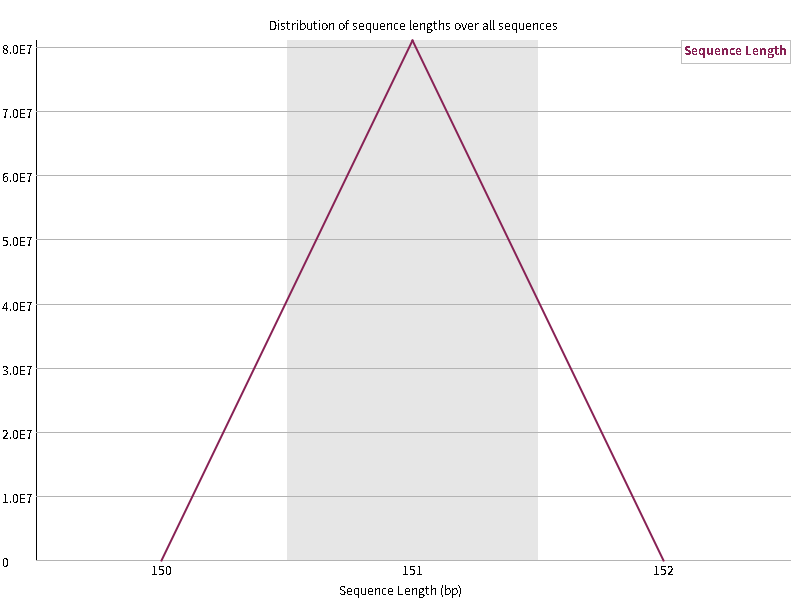
8. Sequence Length Distribution
每次测序仪测出来的长度在理论上应该是完全相等的,但是总会有一些偏差,如此图中,151bp是主要的,但是还是有少量的150和152bp的长度,不过数量比较少,不影响后续分析,当测序的长度有很大不同时,则表明测序仪在此次测序过程中产生的数据不可信,但对于某些测序平台,具有不同的read长度是完全正常的。当reads长度不一致时警告,当有长度为0的read时不合格。
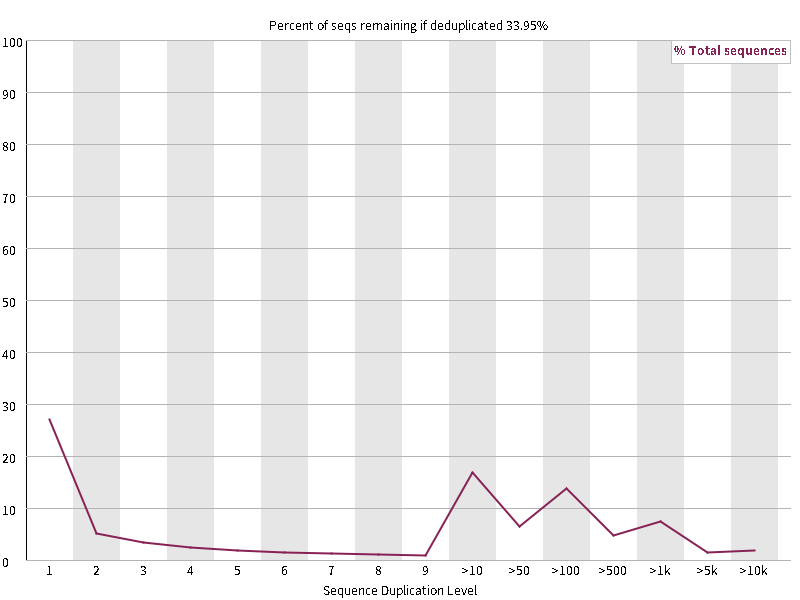
9. Sequence Duplication Levels
横轴为reads重复的次数,纵轴为重复次数对应的reads占不重复的reads的比例。测序深度越高,越容易产生一定程度的duplication,这是正常的现象,但如果duplication的程度很高,那么表明存在富集的偏好(enrichment bias)(比如:测序过程中的PCR重复,转录组测序中某些基因表达量高),序列重复比例越高,则表明实际有用的序列越少。图中有蓝红两条线,蓝色线表示的是文件中所有的序列中duplicate程度的分布,红色线表示的是去冗余之后的序列,含量表示的在全部序列都考虑时不同冗余程度的序列所占的比例。重复reads占总数的比例大于20%时警报,大于50%时不合格。
10. Overrepresented sequences
如果有某个序列大量出现,就叫做over-represented。标准是占全部reads的0.1%以上。但是因为用的是Duplicate sequences前200,000条数据,所以有可能over-represented reads不在里面,参考意义不大。
11. Adapter Content
此图衡量的是序列中两端adapter的情况,如果在fastqc分析的时候-a(指定含adapters序列文件)选项没有内容,则默认使用图例中的通用adapter序列进行统计。含有adapter超过所有reads的5%的警告,超过10%不合格。
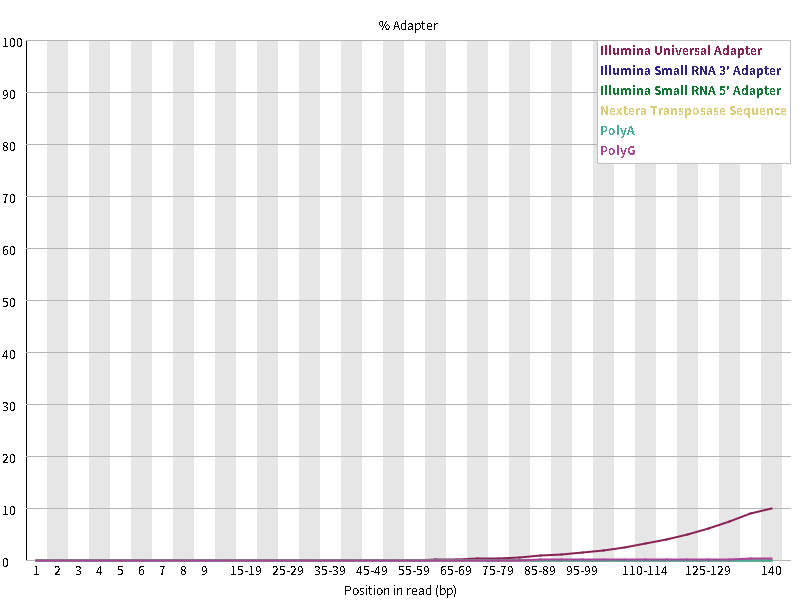
以我这个测序数据为例,主要的问题是含有大量的接头序列,并且reads的前1-15位置的碱基不是很稳定,后面要着重处理。
Fastp对原始数据进行过滤
当我们通过FastQC对测序数据质检,知道数据有哪些错误后,就可以通过fastp和trimmomatic等过滤软件进行针对性的质控,这里我们选择fastp。
安装
mamba install bioconda::fastp
过滤
fastp -i fastp_P636_YMiao_A01v1_S0-1_S1_L002_R1_001_fastqc.html \
-I fastp_P636_YMiao_A01v1_S0-1_S1_L002_R2_001_fastqc.html \
-o fastp_fastp_P636_YMiao_A01v1_S0-1_S1_L002_R1_001_fastqc.html \
-O fastp_fastp_P636_YMiao_A01v1_S0-1_S1_L002_R2_001_fastqc.html \
-t 8 \
-f 15 \
-5
# -i,-i 输入的测序数据
# -o,-O 输出的过滤后
测序数据
# -t cpu
# -f 删除reads头部前15个碱基