1. 前言
无
2. WGCNA分析
准备三个输入文件,(1)表达量矩阵(2)性状数据(3)基因注释信息(可选)
| control1 | control2 | control3 | treat1 | treat2 | treat3 | |
|---|---|---|---|---|---|---|
| evm.model.scaffold_1.4 | 0 | 0.423025 | 1.333393 | 0.084236 | 0 | 0 |
| evm.model.scaffold_1.6 | 70.603968 | 71.632693 | 71.984594 | 84.994817 | 102.021105 | 102.137433 |
| evm.model.scaffold_1.7.1.68d7552f | 11.779294 | 17.393872 | 12.17891 | 5.424567 | 7.394926 | 4.955899 |
| evm.model.scaffold_1.11 | 0 | 0 | 0 | 0 | 0 | 0 |
| evm.model.scaffold_1.12 | 23.808061 | 28.144729 | 18.649506 | 16.886912 | 15.417194 | 12.478677 |
| Stem_diameter | Cross_sectional_area | |
|---|---|---|
| control1 | 4.5 | 14.9 |
| control2 | 3.9 | 11.6 |
| control3 | 4.3 | 14.3 |
| treat1 | 6.1 | 28.7 |
| treat2 | 6.5 | 33.1 |
| treat3 | 6.4 | 31.8 |
| gene_id | gene_symbol | gene_name |
|---|---|---|
| evm.model.scaffold_17.1908 | GERD | GERD |
| evm.model.scaffold_27.5 | truB | truB |
| evm.model.scaffold_27.133 | GSK3B | GSK3B |
| evm.model.scaffold_27.165 | speD | speD |
| evm.model.scaffold_27.198 | E1.14.17.4 | E1.14.17.4 |
| evm.model.scaffold_27.230 | ppa | ppa |
| evm.model.scaffold_19.719 | SLC2A13 | SLC2A13 |
| evm.model.scaffold_19.751 | tag | tag |
| evm.model.scaffold_19.783 | SLC15A3_4 | SLC15A3_4 |
| evm.model.scaffold_19.847 | RP-L6e | RP-L6e |
| evm.model.scaffold_43.781 | REEP5_6 | REEP5_6 |
| evm.model.scaffold_43.877 | E2.1.1.104,ROMT | E2.1.1.104,ROMT |
| evm.model.scaffold_69.1113 | GST | GST |
| evm.model.scaffold_69.1241 | UXS1 | UXS1 |
| evm.model.scaffold_69.1305 | COPS2 | COPS2 |
| evm.model.scaffold_69.1337 | HPAT | HPAT |
library(WGCNA)
library(tidyverse)
gene_exp <- read.table("WGCNA_input_matrix.txt",
row.names = 1)
sample_info <- read.table(file = 'WGCNA_input_triat.txt',
row.names = 1)
gene_info <- read.table(file = 'WGCNA_input_anno.txt', header = TRUE)
## -----------------------------------------------------------
{
# 转置
datExpr0 <- t(gene_exp)
# 缺失数据及无波动数据过滤
gsg <- goodSamplesGenes(
datExpr0,
minFraction = 1/2 #基因的缺失数据比例阈值
)
datExpr <- datExpr0[gsg$goodSamples, gsg$goodGenes]
# 通过聚类,查看是否有明显异常样本, 如果有需要剔除掉
plot(hclust(dist(datExpr)),
cex.lab = 1.5,
cex.axis = 1.5,
cex.main = 2)
# 如果剩余基因仍然太多(如 5 到10万条),
##而电脑配置内存不够,可以进一步过滤掉
##波动小的基因。不要只用差异基因做。
# library(genefilter)
# var_gene_exp <- varFilter(
# as.matrix(t(datExpr)),
# var.func = IQR,
# var.cutoff = 0.5, # 实际项目中设置 0.5 以下
# filterByQuantile = TRUE)
#
# datExpr <- t(var_gene_exp)
datExpr[1:4,1:4]
}
## -----------------------------------------------------------
{
datTraits <- sample_info
datTraits[1:4, 1:4]
}
## ----message=FALSE------------------------------------------
{
library(WGCNA)
# 多线程
enableWGCNAThreads(nThreads = 6)
## disableWGCNAThreads()
# 通过对 power 的多次迭代,确定最佳 power
sft <- pickSoftThreshold(
datExpr,
powerVector = 1:20, # 尝试 1 到 20
networkType = "unsigned"
)
}
## -----------------------------------------------------------
{
# 画图
library(ggplot2)
library(ggrepel)
library(cowplot)
library(ggthemes)
fig_power1 <- ggplot(data = sft$fitIndices,
aes(x = Power,
y = SFT.R.sq)) +
geom_point(color = 'red') +
geom_text_repel(aes(label = Power)) +
geom_hline(aes(yintercept = 0.85), color = 'red') +
labs(title = 'Scale independence',
x = 'Soft Threshold (power)',
y = 'Scale Free Topology Model Fit,signed R^2') +
theme_few() +
theme(plot.title = element_text(hjust = 0.5))
fig_power2 <- ggplot(data = sft$fitIndices,
aes(x = Power,
y = mean.k.)) +
geom_point(color = 'red') +
geom_text_repel(aes(label = Power)) +
labs(title = 'Mean connectivity',
x = 'Soft Threshold (power)',
y = 'Mean Connectivity') +
theme_few()+
theme(plot.title = element_text(hjust = 0.5))
plot_grid(fig_power1, fig_power2)
}
## ----message=FALSE------------------------------------------
{
net <- blockwiseModules(
# 0.输入数据
datExpr,
# 1. 计算相关系数
corType = "pearson", # 相关系数算法,pearson|bicor
# 2. 计算邻接矩阵
power = 12, # 前面得到的 soft power
networkType = "unsigned", # unsigned | signed | signed hybrid
# 3. 计算 TOM 矩阵
TOMType = "unsigned", # none | unsigned | signed
saveTOMs = TRUE,
saveTOMFileBase = "blockwiseTOM",
# 4. 聚类并划分模块
deepSplit = 2, # 0|1|2|3|4, 值越大得到的模块就越多越小
minModuleSize = 30,
# 5. 合并相似模块
## 5.1 计算模块特征向量(module eigengenes, MEs),即 PC1
## 5.2 计算 MEs 与 datTrait 之间的相关性
## 5.3 对距离小于 mergeCutHeight (1-cor)的模块进行合并
mergeCutHeight = 0.25,
# 其他参数
numericLabels = FALSE, # 以数字命名模块
nThreads = 0, # 0 则使用所有可用线程
maxBlockSize = 100000 # 需要大于基因的数目
)
# 查看每个模块包含基因数目
table(net$colors)
}
## -----------------------------------------------------------
{
library(tidyverse)
wgcna_result <- data.frame(gene_id = names(net$colors),
module = net$colors) %>%
left_join(gene_info, by = c('gene_id' = 'gene_id'))
head(wgcna_result)
}
## -----------------------------------------------------------
{
# 同时绘制聚类图和模块颜色
plotDendroAndColors(
dendro = net$dendrograms[[1]],
colors = net$colors,
groupLabels = "Module colors",
dendroLabels = FALSE,
addGuide = TRUE)
}
## ----message=FALSE------------------------------------------
{
# 计算相关性
moduleTraitCor <- cor(
net$MEs,
datTraits,
use = "p",
method = 'spearman' # 注意相关系数计算方式
)
# 计算 Pvalue
moduleTraitPvalue <- corPvalueStudent(
moduleTraitCor,
nrow(datExpr))
}
## -----------------------------------------------------------
{
# 相关性 heatmap
sizeGrWindow(10,6)
# 连接相关性和 pvalue
textMatrix <- paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep = "");
dim(textMatrix) <- dim(moduleTraitCor)
# heatmap 画图
par(mar = c(6, 8.5, 3, 3))
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(net$MEs),
ySymbols = names(net$MEs),
colorLabels = FALSE,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
}
## -----------------------------------------------------------
{
MET <- orderMEs(cbind(net$MEs, dplyr::select(datTraits, Lignin_content)))
plotEigengeneNetworks(
multiME = MET,
setLabels = "Eigengene dendrogram",
plotDendrograms = TRUE,
plotHeatmaps = FALSE,
colorLabels = TRUE,
marHeatmap = c(3,4,2,2))
}
## -----------------------------------------------------------
{
plotEigengeneNetworks(
multiME = MET,
setLabels = "Eigengene dendrogram",
plotDendrograms = FALSE,
plotHeatmaps = TRUE,
colorLabels = TRUE,
marHeatmap = c(8,8,2,2))
}
## -----------------------------------------------------------
{
# 需要导出的模块
my_modules <- c('blue')
# 提取该模块的表达矩阵
m_wgcna_result <- filter(wgcna_result, module %in% my_modules)
m_datExpr <- datExpr[, m_wgcna_result$gene_id]
# 计算该模块的 TOM 矩阵
m_TOM <- TOMsimilarityFromExpr(
m_datExpr,
power = 6,
networkType = "unsigned",
TOMType = "unsigned")
dimnames(m_TOM) <- list(colnames(m_datExpr), colnames(m_datExpr))
# 导出 Cytoscape 输入文件
cyt <- exportNetworkToCytoscape(
m_TOM,
edgeFile = "CytoscapeInput-network.txt",
weighted = TRUE,
threshold = 0.2)
}
# 导出指定模块的基因
module_genes <- m_wgcna_result$gene_id
write.table(
module_genes,
file = "blue_module_all_genes.txt",
quote = FALSE,
row.names = FALSE,
col.names = FALSE
)
# 导出hub基因的自网络
kWithin <- rowSums(m_TOM) - 1
summary(kWithin)
ME_name <- "MEblue"
MM <- cor(m_datExpr, net$MEs[, ME_name], use = "p")
summary(abs(MM))
top_frac <- 0.1 # 5%
top_n <- ceiling(length(kWithin) * top_frac)
hub_genes <- names(sort(kWithin, decreasing = TRUE))[1:top_n]
length(hub_genes)
hub_TOM <- m_TOM[hub_genes, hub_genes]
exportNetworkToCytoscape(
hub_TOM,
edgeFile = "blue_hub_network.txt",
weighted = TRUE,
threshold = 0.1,
nodeNames = hub_genes
)
hub_node_df <- data.frame(
gene_id = hub_genes,
module = "blue",
kWithin = kWithin[hub_genes],
MM = MM[hub_genes]
) %>%
left_join(gene_info, by = "gene_id")
write.table(
hub_node_df,
file = "blue_hub_nodes.txt",
sep = "\t",
quote = FALSE,
row.names = FALSE
)
# 导出指定基因集的子网络
## ===============================
## 边稀疏化函数:每个节点只保留 top-k strongest edges
## ===============================
sparsify_TOM_topK <- function(TOM, k = 5) {
keep <- matrix(FALSE, nrow = nrow(TOM), ncol = ncol(TOM))
rownames(keep) <- colnames(keep) <- colnames(TOM)
for (g in rownames(TOM)) {
v <- TOM[g, ]
v <- sort(v, decreasing = TRUE)
nbrs <- names(v)[2:(k + 1)] # 第 1 个是自己
keep[g, nbrs] <- TRUE
}
# 保证无向图对称
keep <- keep | t(keep)
TOM_sparse <- TOM
TOM_sparse[!keep] <- 0
diag(TOM_sparse) <- 0
return(TOM_sparse)
}
## ===============================
## 参数区(你通常只需要改这里)
## ===============================
target_file <- "blue_model_muzhisu.gene" # 你的 txt 文件
k_per_gene <- 30 # 每个目标基因取多少邻居
max_nodes <- 150 # 子网络最大节点数(自动控制)
threshold <- 0.05 # TOM 边阈值
## ===============================
## Step 1. 读取目标基因列表
## ===============================
target_genes_raw <- read.table(
target_file,
header = TRUE,
stringsAsFactors = FALSE
)
target_genes <- unique(target_genes_raw[[1]])
## ===============================
## Step 2. 仅保留 blue 模块中、且存在于 TOM 的基因
## ===============================
target_genes <- intersect(
target_genes,
colnames(m_TOM)
)
if (length(target_genes) == 0) {
stop("❌ None of the target genes are found in the blue module TOM matrix.")
}
message("✔ Target genes used: ", length(target_genes))
## ===============================
## Step 3. 为每个目标基因提取 Top-k 邻居
## ===============================
neighbor_list <- lapply(target_genes, function(g) {
tom_vec <- m_TOM[g, ]
tom_vec <- sort(tom_vec, decreasing = TRUE)
names(tom_vec)[2:(k_per_gene + 1)] # 去掉自己
})
names(neighbor_list) <- target_genes
## ===============================
## Step 4. 合并所有 ego-network(方案 2 核心)
## ===============================
ego_union <- unique(c(target_genes, unlist(neighbor_list)))
message("✔ Initial subnetwork size: ", length(ego_union))
## ===============================
## Step 5. 自动控制网络规模(防止过大)
## 原则:优先保留目标基因 + 与目标基因 TOM 总和高的节点
## ===============================
if (length(ego_union) > max_nodes) {
# 计算每个节点与所有 target_genes 的 TOM 总和
tom_score <- sapply(ego_union, function(g) {
sum(m_TOM[g, target_genes], na.rm = TRUE)
})
ego_union <- names(sort(tom_score, decreasing = TRUE))[1:max_nodes]
message("✔ Subnetwork trimmed to max_nodes = ", max_nodes)
}
## ===============================
## Step 6. 提取子 TOM 并导出 Cytoscape edge 文件
## ===============================
sub_TOM <- m_TOM[ego_union, ego_union]
# ⭐ 每个节点只保留 strongest 的 5 条边
sub_TOM_sparse <- sparsify_TOM_topK(sub_TOM, k = 5)
exportNetworkToCytoscape(
sub_TOM_sparse,
edgeFile = "blue_targetGene_subnetwork_edges.txt",
weighted = TRUE,
threshold = 0.001, # 非常低,仅用于过滤 0
nodeNames = ego_union
)
## ===============================
## Step 7. 构建并导出带注释的 node 文件(强烈推荐)
## ===============================
node_df <- data.frame(
gene_id = ego_union,
is_target = ego_union %in% target_genes
) %>%
left_join(wgcna_result, by = "gene_id") %>% # module + 可能已有注释
left_join(gene_info, by = "gene_id") # 你的注释文件
write.table(
node_df,
file = "blue_targetGene_subnetwork_nodes.txt",
sep = "\t",
quote = FALSE,
row.names = FALSE
)
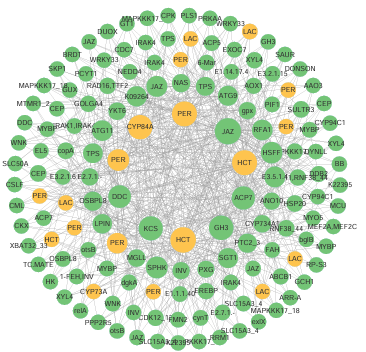
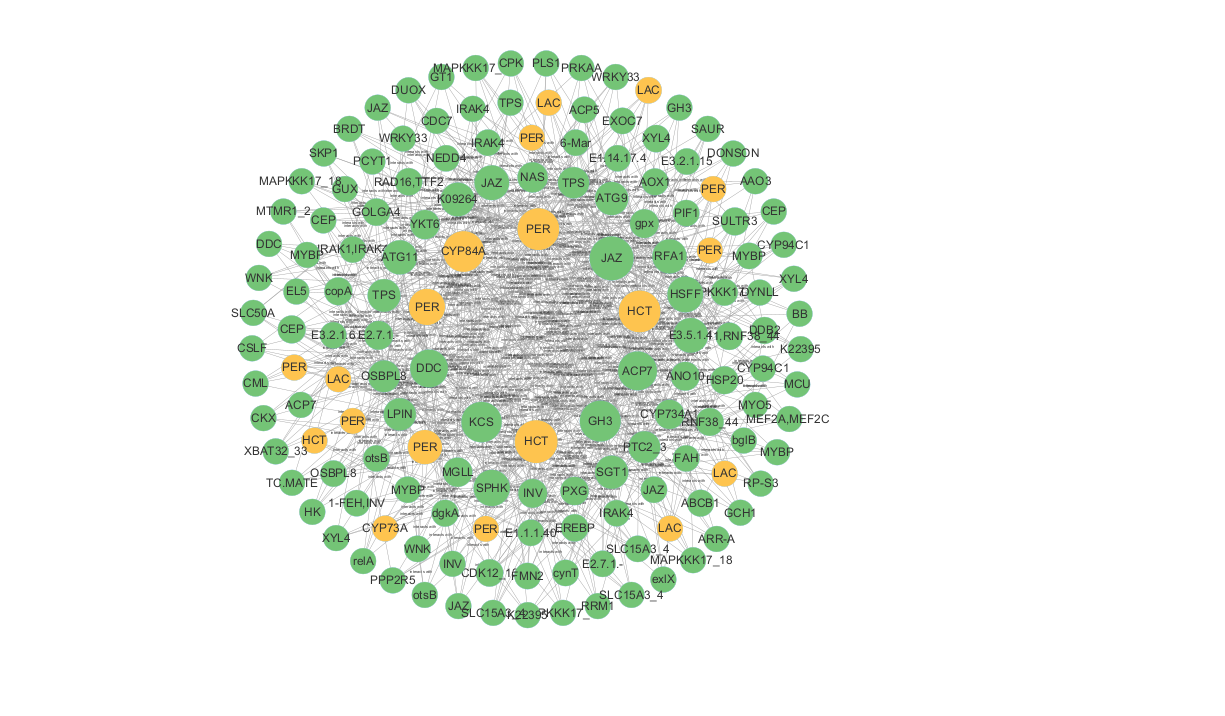
随后把node和edge文件导入cytoscape进行可视化即可。