1. 前言
比较转录组作为生信分析中最常规的分析,底层逻辑是通过基因的表达量,筛选差异基因,随后进行下游的富集分析。也可以通过表达量矩阵进行时序分析和WGCNA分析。那么,这种分析模式可不可以用在其他分析中呢,我个人认为是可以的。因此今天就把这套流程应用在宏基因组分析中,用基因的丰度代替基因的表达量,来进行一些分析。
2. 分析
2.1 DESeq2差异分析
通过基因的丰度数据进行差异分析,需要注释的是,丰度要是原始的Count,不能是标准化后的TMP和FPKM。这一步的输入文件是(1)基因丰度(2)样品分组信息
| sample | condition |
|---|---|
| J1 | J |
| J2 | J |
| J3 | J |
| J4 | J |
| F1 | F |
| F2 | F |
| F3 | F |
| F4 | F |
| GeneID | F2 | J1 | J4 | J3 | J2 | F4 | F1 | F3 | Total |
|---|---|---|---|---|---|---|---|---|---|
| J2_k97_737169_1 | 298 | 318 | 4 | 2 | 8 | 90 | 20 | 30 | 770 |
| J2_k97_737170_1 | 0 | 0 | 2 | 2 | 6 | 0 | 0 | 0 | 10 |
| J2_k97_105311_1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 0 | 4 |
| J2_k97_1684952_1 | 0 | 0 | 8 | 0 | 6 | 0 | 0 | 0 | 14 |
| J2_k97_1369023_1 | 0 | 0 | 2 | 0 | 2 | 0 | 0 | 2 | 6 |
| J2_k97_1369023_2 | 0 | 0 | 2 | 4 | 10 | 0 | 0 | 2 | 18 |
| J2_k97_631861_1 | 0 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 2 |
# --- 1. 设置工作环境 ---
# 加载所需的R包
# 如果您尚未安装这些包,请先运行 install.packages("包名") 进行安装
# 例如: install.packages("DESeq2")
suppressPackageStartupMessages({
library(DESeq2)
library(readr)
library(dplyr)
})
# --- 2. 设置输入和输出文件 ---
# 请根据您的文件位置修改以下路径
# 设置原始基因count矩阵文件的路径 (例如: "data/reads_number.xls")
# 文件应为制表符分隔,第一列为基因ID,后续列为样本的count值。
count_file <- "reads_number.xls"
# 设置样本信息文件的路径 (例如: "data/sample_info.csv")
# 文件应为逗号分隔,包含两列: "sample" (样本名称) 和 "condition" (分组信息)。
sample_info_file <- "sample_info.csv"
# 设置输出文件的前缀 (例如: "results/deseq2_output")
# 脚本将生成一个名为 <output_prefix>_diff_expression.csv 的结果文件。
output_prefix <- "deseq2_results"
# --- 3. 读取并预处理数据 ---
# 读取原始基因count矩阵
# 假设文件是制表符分隔的,第一列是基因ID
cat(paste0("正在读取count文件: ", count_file, "\n"))
count_data <- read.delim(count_file, row.names = 1, sep = "\t", check.names = FALSE)
# 删除count数据中的最后一列
# 原始脚本位置: /home/majunpeng/sda2/Lishuyan_Kuozangzi_sequence/Metagenome/Gemini/deseq2_analysis_rstudio_1.R
count_data <- count_data[, -ncol(count_data)]
# 读取样本信息文件
# 假设文件是逗号分隔的
cat(paste0("正在读取样本信息文件: ", sample_info_file, "\n"))
sample_info <- read_csv(sample_info_file, show_col_types = FALSE)
# 确保count数据是整数类型,DESeq2要求输入为整数
count_data <- round(count_data)
# 确保样本信息中的样本名称与count数据中的列名一致且顺序一致
# 筛选出在count数据中存在的样本
sample_info <- sample_info %>%
filter(sample %in% colnames(count_data)) %>%
arrange(match(sample, colnames(count_data))) # 按照count数据的列顺序排序
# 检查样本数量是否匹配
if (ncol(count_data) != nrow(sample_info)) {
stop("错误: count数据中的样本列数与样本信息文件中的样本行数不匹配。请检查您的输入文件。\n")
}
# 确保count数据的列名和sample_info的sample列顺序一致
count_data <- count_data[, sample_info$sample]
# 将condition列转换为因子,并设置比较组和对照组
# DESeq2默认按字母顺序确定对照组,例如'F'和'J'中,'F'会成为对照组。
# 为了明确指定对照组,我们使用relevel()函数。
# 这里我们将'J'设置为对照组(reference level)。
unique_conditions <- unique(sample_info$condition)
if (length(unique_conditions) < 2) {
stop("错误: 样本信息中至少需要两个不同的条件才能进行差异表达分析。\n")
}
# 假设您想比较 F组 vs J组,J组是对照组
sample_info$condition <- factor(sample_info$condition)
sample_info$condition <- relevel(sample_info$condition, ref = "F")
# --- 4. 构建DESeqDataSet对象 ---
# 创建DESeqDataSet对象
# design公式指定了如何进行差异分析,这里是根据condition进行比较
dds <- DESeqDataSetFromMatrix(countData = count_data,
colData = sample_info,
design = ~ condition)
# 过滤低表达基因 (可选,但推荐)
# 过滤掉在所有样本中count总和过低的基因,以减少计算量和提高统计功效
# 这里保留那些在至少一个样本中count大于等于10的基因
keep <- rowSums(counts(dds) >= 10) >= 1
dds <- dds[keep,]
# --- 5. 运行DESeq2核心分析 ---
cat("正在运行DESeq2差异表达分析...\n")
dds <- DESeq(dds)
# --- 6. 提取和保存结果 ---
# 提取差异表达结果
# resultsNames(dds) 可以查看所有可用的比较名称
# 默认情况下,它会比较处理组与您设定的对照组
res <- results(dds)
# 您也可以使用 contrast 参数明确指定比较,例如比较'F'组相对于'J'组的差异
# res <- results(dds, contrast = c("condition", "F", "J"))
# 对结果进行排序 (例如,按调整后的p值(padj)从小到大排序)
res_ordered <- res[order(res$padj),]
# 将结果转换为数据框
res_df <- as.data.frame(res_ordered)
# 添加基因ID列
res_df <- tibble::rownames_to_column(res_df, var = "gene_id")
# 保存结果到CSV文件
output_file <- paste0(output_prefix, "_diff_expression.csv")
cat(paste0("正在保存差异表达结果到: ", output_file, "\n"))
write_csv(res_df, output_file)
cat("DESeq2分析完成!\n")
最后,我们就一定拿到了差异分析的结果,接下来我们以P值小于等于0.05,Log2FC的绝对值大于等于1.5进行差异基因的筛选。
| gene_id | baseMean | log2FoldChange | lfcSE | stat | pvalue | padj |
|---|---|---|---|---|---|---|
| J3_k97_1869542_48 | 719.9416276 | 26.53634325 | 2.584192382 | 10.26871816 | 9.75E-25 | 1.10E-18 |
| J3_k97_1869542_49 | 505.5442894 | 26.1670583 | 2.571273542 | 10.17669177 | 2.52E-24 | 1.42E-18 |
| J3_k97_1869542_3 | 502.7114513 | 26.08115486 | 2.581835499 | 10.101788 | 5.42E-24 | 1.53E-18 |
| J3_k97_1869542_47 | 495.3948077 | 25.74264487 | 2.546306163 | 10.10979954 | 5.00E-24 | 1.53E-18 |
| J3_k97_1869542_33 | 623.4655207 | 26.26834756 | 2.607327384 | 10.0748175 | 7.14E-24 | 1.61E-18 |
| J3_k97_1869542_67 | 311.7707339 | 25.4921083 | 2.547029189 | 10.00856543 | 1.40E-23 | 2.63E-18 |
| J3_k97_1869542_32 | 554.8218292 | 26.2904605 | 2.632630124 | 9.986385957 | 1.75E-23 | 2.82E-18 |
| J3_k97_1869542_14 | 377.7311327 | 25.44960062 | 2.552398401 | 9.970857455 | 2.04E-23 | 2.89E-18 |
| J3_k97_1869542_8 | 404.1818614 | 25.85406933 | 2.612079894 | 9.89788612 | 4.25E-23 | 4.23E-18 |
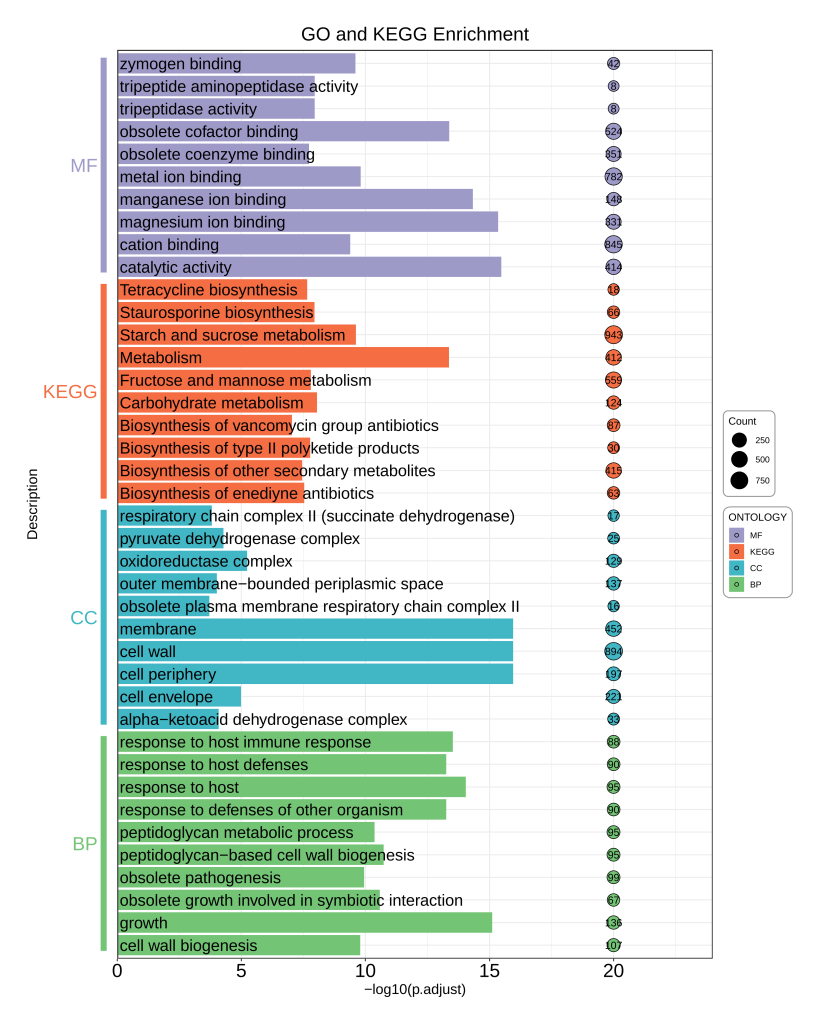
2.2 富集分析
富集分析主要分为两部分,常规的KEGG和GO富集分析和GSEA富集分析。第一步就是要通过Eggnog的注释结果构建一个Orgdb库。
emapper
Rscript /pub/software/emcp/emapperx.R out.emapper.annotations proteins.fa
随后进行常规的KEGG和GO富集分析,这一步我使用的是TBtools,在这里就不详细解释怎么用了,请自行搜索学习。完成富集分析后,按照下面的输入文件格式简单的整理一下,下面的是一个KEGG和GO富集可视化的代码。
| ONTOLOGY | Description | p.adjust | Count |
|---|---|---|---|
| KEGG | Metabolism | 4.30E-14 | 412 |
| KEGG | Starch and sucrose metabolism | 2.42E-10 | 943 |
| KEGG | Carbohydrate metabolism | 8.78E-09 | 124 |
| KEGG | Staurosporine biosynthesis | 1.11E-08 | 66 |
| KEGG | Fructose and mannose metabolism | 1.56E-08 | 559 |
| KEGG | Biosynthesis of type II polyketide products | 1.68E-08 | 30 |
| KEGG | Tetracycline biosynthesis | 2.21E-08 | 18 |
| KEGG | Biosynthesis of enediyne antibiotics | 2.91E-08 | 63 |
| KEGG | Biosynthesis of other secondary metabolites | 3.49E-08 | 415 |
| KEGG | Biosynthesis of vancomycin group antibiotics | 9.06E-08 | 87 |
| MF | catalytic activity | 3.33E-16 | 414 |
| MF | magnesium ion binding | 4.44E-16 | 331 |
| MF | manganese ion binding | 4.66E-15 | 148 |
| MF | obsolete cofactor binding | 4.13E-14 | 524 |
| MF | metal ion binding | 1.55E-10 | 782 |
| MF | zymogen binding | 2.49E-10 | 42 |
| MF | cation binding | 4.05E-10 | 845 |
| MF | tripeptide aminopeptidase activity | 1.10E-08 | 8 |
| MF | tripeptidase activity | 1.10E-08 | 8 |
| MF | obsolete coenzyme binding | 1.84E-08 | 351 |
| CC | cell periphery | 1.11E-16 | 197 |
| CC | cell wall | 1.11E-16 | 894 |
| CC | membrane | 1.11E-16 | 452 |
| CC | oxidoreductase complex | 5.77E-06 | 129 |
| CC | cell envelope | 1.03E-05 | 221 |
| CC | pyruvate dehydrogenase complex | 5.25E-05 | 25 |
| CC | alpha-ketoacid dehydrogenase complex | 8.13E-05 | 33 |
| CC | outer membrane-bounded periplasmic space | 9.59E-05 | 137 |
| CC | respiratory chain complex II (succinate dehydrogenase) | 0.000151272 | 17 |
| CC | obsolete plasma membrane respiratory chain complex II | 0.000192509 | 16 |
| BP | growth | 7.77E-16 | 136 |
| BP | response to host | 8.88E-15 | 95 |
| BP | response to host immune response | 2.95E-14 | 88 |
| BP | response to defenses of other organism | 5.51E-14 | 90 |
| BP | response to host defenses | 5.51E-14 | 90 |
| BP | peptidoglycan-based cell wall biogenesis | 1.84E-11 | 95 |
| BP | obsolete growth involved in symbiotic interaction | 2.59E-11 | 67 |
| BP | peptidoglycan metabolic process | 4.20E-11 | 95 |
| BP | obsolete pathogenesis | 1.12E-10 | 99 |
| BP | cell wall biogenesis | 1.61E-10 | 107 |
rm(list = ls())
####----load R Package----####
library(tidyverse)
library(ggh4x)
library(ggfun)
library(clusterProfiler)
library(org.Hs.eg.db)
####----load Result----####
plot_df <- read.csv("KEGG_GO富集绘图.csv")
####----Plot----####
plot <- plot_df %>%
ggplot() +
geom_bar(aes(x = -log10(p.adjust), y = interaction(Description, ONTOLOGY), fill = ONTOLOGY), stat = "identity") +
geom_text(aes(x = 0.1, y = interaction(Description, ONTOLOGY), label = Description), size = 6, hjust = "left", color = "#000000") +
geom_point(aes(x = 20, y = interaction(Description, ONTOLOGY), size = Count, fill = ONTOLOGY), shape = 21) +
geom_text(aes(x = 20, y = interaction(Description, ONTOLOGY), label = Count)) +
scale_x_continuous(expand = expansion(mult = c(0, 0.2))) +
scale_fill_manual(values = c("#74c476", "#41b6c4", "#f46d43", "#9e9ac8")) +
scale_size(range = c(5, 8.5),
guide = guide_legend(override.aes = list(fill = "#000000"))) +
guides(y = "axis_nested",
fill = guide_legend(reverse = T)) +
labs(x = "-log10(p.adjust)", y = "Description") +
ggtitle(label = "GO and KEGG Enrichment") +
theme_bw() +
theme(
ggh4x.axis.nestline.y = element_line(size = 3, color = c("#74c476", "#41b6c4", "#f46d43", "#9e9ac8")),
ggh4x.axis.nesttext.y = element_text(colour = c("#74c476", "#41b6c4", "#f46d43", "#9e9ac8")),
legend.background = element_roundrect(color = "#969696"),
panel.border = element_rect(linewidth = 1),
plot.margin = margin(t = 1, r = 1, b = 1, l = 1, unit = "cm"),
axis.text = element_text(color = "#000000", size = 20),
axis.text.y = element_text(color = rep(c("#41ae76", "#225ea8", "#fc4e2a", "#88419d"), each = 10)),
axis.text.y.left = element_blank(),
axis.ticks.length.y.left = unit(10, "pt"),
axis.ticks.y.left = element_line(color = NA),
axis.title = element_text(color = "#000000", size = 15),
plot.title = element_text(color = "#000000", size = 20, hjust = 0.5)
)
plot
ggsave(filename = "GO_KEGG_Lishuyan.pdf",
plot = plot,
height = 15,
width = 12)
####----sessionInfo----####
sessionInfo()
| gene_id | log2FoldChange |
|---|---|
| J3_k97_1869542_48_1 | 26.53634325 |
| J3_k97_1869542_32_1 | 26.2904605 |
| J3_k97_1869542_33_1 | 26.26834756 |
| J3_k97_1869542_49_1 | 26.1670583 |
| J3_k97_1869542_3_1 | 26.08115486 |
| J3_k97_1869542_45_1 | 25.94958501 |
| J3_k97_1869542_8_1 | 25.85406933 |
| J3_k97_1869542_23_1 | 25.82508916 |
| J3_k97_1869542_47_1 | 25.74264487 |
| J3_k97_1869542_75_1 | 25.69997138 |
| J3_k97_1869542_60_1 | 25.6210279 |
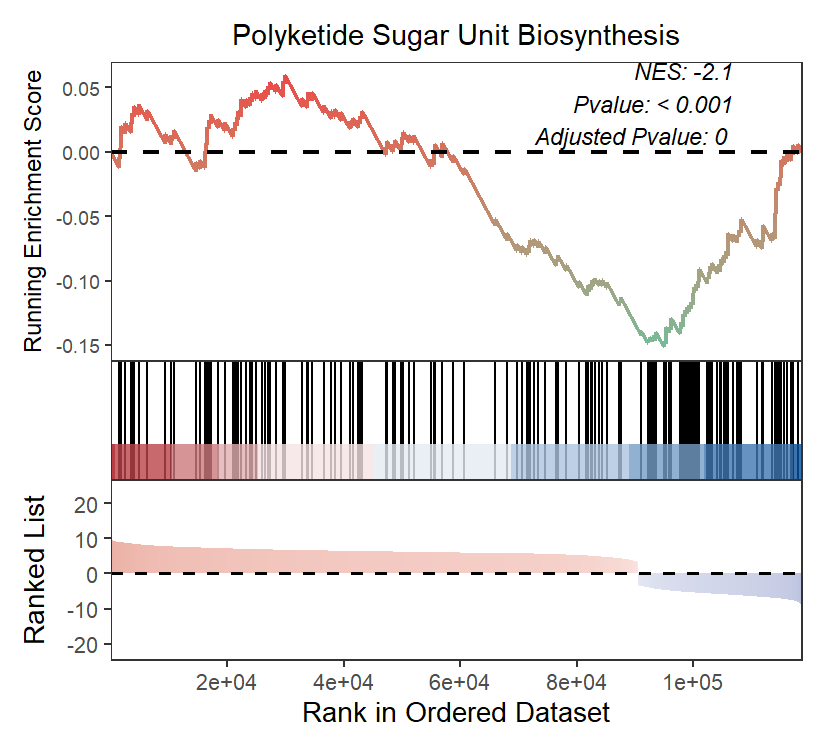
接下来进行GSEA富集分析,同样,需要按照下面的格式整理的一个输入文件,包含基因ID和Log2FC值信息。
library(magrittr)
library(readr)
library(clusterProfiler)
library(tidyr)
library(tibble)
library(stringr)
library(GseaVis)
get_path2name <- function(){
keggpathid2name.df <- clusterProfiler:::kegg_list("pathway")
keggpathid2name.df[,1] %<>% gsub("path:map", "", .)
colnames(keggpathid2name.df) <- c("path_id","path_name")
return(keggpathid2name.df)
}
pathway2name <- get_path2name()
pathway2name$path_id <- gsub("map", "", pathway2name$path_id, ignore.case = TRUE)
emapper <- read_delim("../EGGNOG.emapper.annotations",
"\t", escape_double = FALSE, col_names = FALSE,
comment = "#", trim_ws = TRUE) %>%
dplyr::select(GID = X1,
COG = X7,
Gene_Name = X8,
Gene_Symbol = X9,
GO = X10,
KO = X12,
Pathway = X13
)
pathway2gene <- dplyr::select(emapper, Pathway, GID) %>%
separate_rows(Pathway, sep = ',', convert = F) %>%
filter(str_detect(Pathway, 'ko')) %>%
mutate(Pathway = str_remove(Pathway, 'ko'))
gene_data <- read.csv("差异基因表达量.csv")
geneList <- setNames(gene_data$log2FoldChange, gene_data$gene_id)
geneList <- sort(geneList, decreasing = TRUE)
head(geneList)
ekp2 = GSEA(
geneList,
TERM2GENE = pathway2gene,
TERM2NAME = pathway2name,
minGSSize = 10,
maxGSSize = 500,
pvalueCutoff = 0.05)
ekp2_df <- as.data.frame(ekp2)
write.csv(as.data.frame(ekp2), "GSEA_analysis_KEGG.csv", row.names = FALSE)
gseaNb(object = ekp2,
geneSetID = '00523',
addPval = T,
rankSeq = 20000)
GSEA_fig <- gseaNb(object = ekp2,
geneSetID = c('00523',"01059", "00404"),
addPval = T,
rankSeq = 20000,
pvalX = 0.15,pvalY = 0.15)
ggsave("GSEA.pdf", GSEA_fig, width = 8, height = 6, dpi = 300)
2.3 WGCNA分析
同样WGCNA也可以做,找到和某个ASV相关性最高的基因。
library(WGCNA)
library(tidyverse)
# 读取基因丰度文件
gene_exp <- read.csv(file = '差异基因_TPM.csv',
row.names = 1)
# 读取扩增子ASV风度文件
sample_info <- read.table(file = 'ASV_Count.txt',
row.names = 1, header = TRUE )
{
# 转置
datExpr0 <- t(gene_exp)
# 缺失数据及无波动数据过滤
gsg <- goodSamplesGenes(
datExpr0,
minFraction = 1/2 #基因的缺失数据比例阈值
)
datExpr <- datExpr0[gsg$goodSamples, gsg$goodGenes]
# 通过聚类,查看是否有明显异常样本, 如果有需要剔除掉
plot(hclust(dist(datExpr)),
cex.lab = 1.5,
cex.axis = 1.5,
cex.main = 2)
# 如果剩余基因仍然太多(如 5 到10万条),
##而电脑配置内存不够,可以进一步过滤掉
##波动小的基因。不要只用差异基因做。
# library(genefilter)
# var_gene_exp <- varFilter(
# as.matrix(t(datExpr)),
# var.func = IQR,
# var.cutoff = 0.5, # 实际项目中设置 0.5 以下
# filterByQuantile = TRUE)
#
# datExpr <- t(var_gene_exp)
datExpr[1:4,1:4]
}
{
datTraits <- sample_info
}
{
library(WGCNA)
# 多线程
enableWGCNAThreads(nThreads = 80)
## disableWGCNAThreads()
# 通过对 power 的多次迭代,确定最佳 power
sft <- pickSoftThreshold(
datExpr,
powerVector = 1:20, # 尝试 1 到 20
networkType = "unsigned"
)
}
{
# 画图
library(ggplot2)
library(ggrepel)
library(cowplot)
library(ggthemes)
fig_power1 <- ggplot(data = sft$fitIndices,
aes(x = Power,
y = SFT.R.sq)) +
geom_point(color = 'red') +
geom_text_repel(aes(label = Power)) +
geom_hline(aes(yintercept = 0.85), color = 'red') +
labs(title = 'Scale independence',
x = 'Soft Threshold (power)',
y = 'Scale Free Topology Model Fit,signed R^2') +
theme_few() +
theme(plot.title = element_text(hjust = 0.5))
fig_power2 <- ggplot(data = sft$fitIndices,
aes(x = Power,
y = mean.k.)) +
geom_point(color = 'red') +
geom_text_repel(aes(label = Power)) +
labs(title = 'Mean connectivity',
x = 'Soft Threshold (power)',
y = 'Mean Connectivity') +
theme_few()+
theme(plot.title = element_text(hjust = 0.5))
plot_grid(fig_power1, fig_power2)
}
{
net <- blockwiseModules(
# 0.输入数据
datExpr,
# 1. 计算相关系数
corType = "pearson", # 相关系数算法,pearson|bicor
# 2. 计算邻接矩阵
power = 6, # 前面得到的 soft power
networkType = "unsigned", # unsigned | signed | signed hybrid
# 3. 计算 TOM 矩阵
TOMType = "unsigned", # none | unsigned | signed
saveTOMs = TRUE,
saveTOMFileBase = "blockwiseTOM",
# 4. 聚类并划分模块
deepSplit = 2, # 0|1|2|3|4, 值越大得到的模块就越多越小
minModuleSize = 30,
# 5. 合并相似模块
## 5.1 计算模块特征向量(module eigengenes, MEs),即 PC1
## 5.2 计算 MEs 与 datTrait 之间的相关性
## 5.3 对距离小于 mergeCutHeight (1-cor)的模块进行合并
mergeCutHeight = 0.25,
# 其他参数
numericLabels = FALSE, # 以数字命名模块
nThreads = 0, # 0 则使用所有可用线程
maxBlockSize = 200000 # 需要大于基因的数目
)
# 查看每个模块包含基因数目
table(net$colors)
}
{
# 同时绘制聚类图和模块颜色
plotDendroAndColors(
dendro = net$dendrograms[[1]],
colors = net$colors,
groupLabels = "Module colors",
dendroLabels = FALSE,
addGuide = TRUE)
}
{
# 计算相关性
moduleTraitCor <- cor(
net$MEs,
datTraits,
use = "p",
method = 'spearman' # 注意相关系数计算方式
)
# 计算 Pvalue
moduleTraitPvalue <- corPvalueStudent(
moduleTraitCor,
nrow(datExpr))
}
{
# 相关性 heatmap
sizeGrWindow(10,6)
# 连接相关性和 pvalue
textMatrix <- paste(signif(moduleTraitCor, 2), "\n(",
signif(moduleTraitPvalue, 1), ")", sep = "");
dim(textMatrix) <- dim(moduleTraitCor)
# heatmap 画图
par(mar = c(6, 8.5, 3, 3))
labeledHeatmap(Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(net$MEs),
ySymbols = names(net$MEs),
colorLabels = FALSE,
colors = blueWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
}
{
# 创建简化版的 WGCNA 结果(不需要 gene_info)
wgcna_result <- data.frame(
gene_id = names(net$colors),
module = net$colors,
stringsAsFactors = FALSE
)
# 查看结果
head(wgcna_result)
table(wgcna_result$module)
}
{
# 需要导出的模块
my_modules <- c('brown')
# 提取该模块的表达矩阵
m_wgcna_result <- filter(wgcna_result, module %in% my_modules)
m_datExpr <- datExpr[, m_wgcna_result$gene_id]
# 计算该模块的 TOM 矩阵
m_TOM <- TOMsimilarityFromExpr(
m_datExpr,
power = 6,
networkType = "unsigned",
TOMType = "unsigned")
dimnames(m_TOM) <- list(colnames(m_datExpr), colnames(m_datExpr))
# 导出 Cytoscape 输入文件
cyt <- exportNetworkToCytoscape(
m_TOM,
edgeFile = "CytoscapeInput-network.txt",
weighted = TRUE,
threshold = 0.2)
}
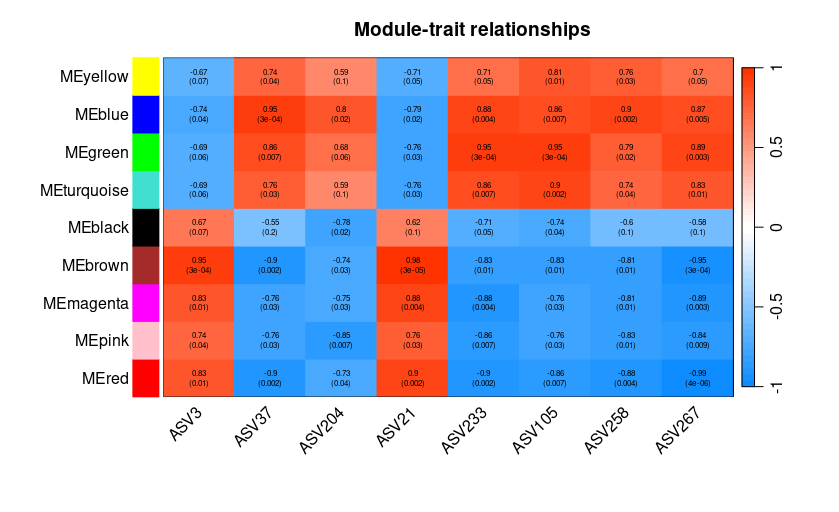
通过每个模块上方显著性和下方的p值来筛选显著的模块,比如和ASV呈现正相关的为brown,负相关的为blue。