0. 前言
去年的时候,我们组想在短时间内快速的出一篇一区的文章,在我的推荐下,老师选择了那时候还在风口浪尖的T2T基因组。我们选了一个基因组预估大小在250M左右,具有一定的经济价值的植物,信心满满的测了基因组(其实我们实现是不知道倍性和基因组大小的,只能通过它的姐妹类群来估算)。当时我们的策略就是HIFI 60,HIC 100,超长 30。
1. 遇到的问题及解决方案
结果等Survey分析结果出来,这个物种的基因组并不是我们预估的二倍体,基因组大小也不是250M,给出的结果是12倍体,基因组大小1.6G。所有人都傻眼了,但是数据都测了,还能怎么办,最后经过我和老师一商量,决定直接用超长的数据组,就算组不到T2T,高倍性也是一个不错的噱头,说干就干,开始组。通过Hifiasm的初步组装,组装大小在1.7G左右,Contig N50为20M,还算符合预期,随后就继续测HIC,准备回来直接挂载。这中间陆陆续续因为其他的课题,这个课题就搁置小半年,最近才开始做。染色体挂载后,我傻眼了,8个染色体,同源染色体的数量各不相同,有5,6,7,8的。我虽然有一些组装基因组的经验,但是如此高倍性的基因组还是第一次碰到,一开始我怀疑可能是把另外一个单倍型的基因组混进来了,于是我开始调整hifiasm的-s参数,但是并没有什么卵用。随后,我把ont数据比对回基因组,如果真的把另外一个单倍型混进来,那所有同源染色体的覆盖度肯定要低于测序深度吗,反之如果少了就会高。结果如下,
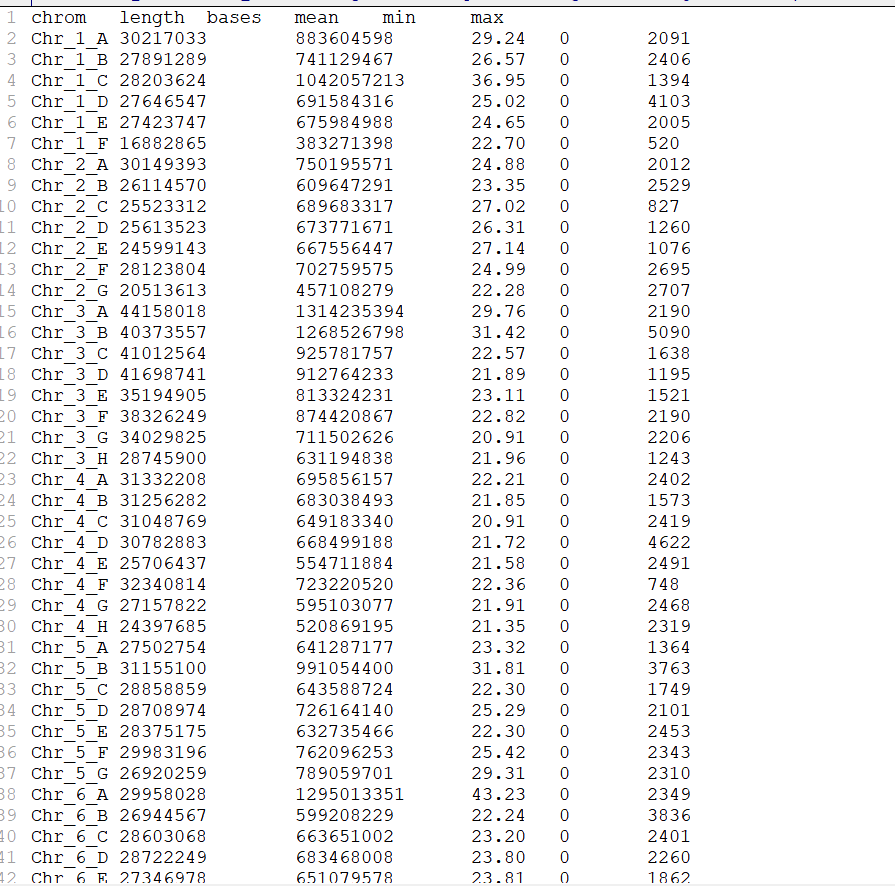
可以清晰的看到,只有存在8个同源染色体的Chr3和Chr4的染色体和平均测序深度20差不多,而Chr1只有6条同源染色体,每条的平均测序深度均明显高于平均的20。到这里,基本上可以判断,这个物种并非是12倍体,而是16倍体,基因组大小应该是2个G。随后,通过咨询曾老师(Haohic的软件作者),得知之所以有的是5,6,7而并非都是8,是因为collapse的存在,翻译过来也就是坍塌区,形成的原因就是同源染色体高度相似,导致组装后完全重合形成了1个。
既然找到问题了,接下来就是解决了,曾老师推荐的方案是,把collapse区域的contig直接复制一个拷贝,相当于是手动的制造一个拷贝,然后挂载上去。这个方案的第一步就是要确定那些区域是collapse区域,可以通过滑窗统计ont测序深度来看那些区域是collapse区域。曾老师的文章中形象生动的解释了什么是collapse(此方法行不通,请自行看下面的加笔)。
minimap2 -t 60 -ax map-ont out_JBAT.FINAL.fa ../../../YY-2.pass.all.fq.gz|samtools view -bS -@ 60 - | samtools sort -@ 60 -o aln.sorted.bam
samtools index aln.sorted.bam
mosdepth -t 10 --by 100000 100000_genome_depth aln.sorted.bam
# plot depth stat result
python plot_depth_along_chromosome.py -i 100000_genome_depth.regions.bed -c scaffold_10 -o scaffold_10.png
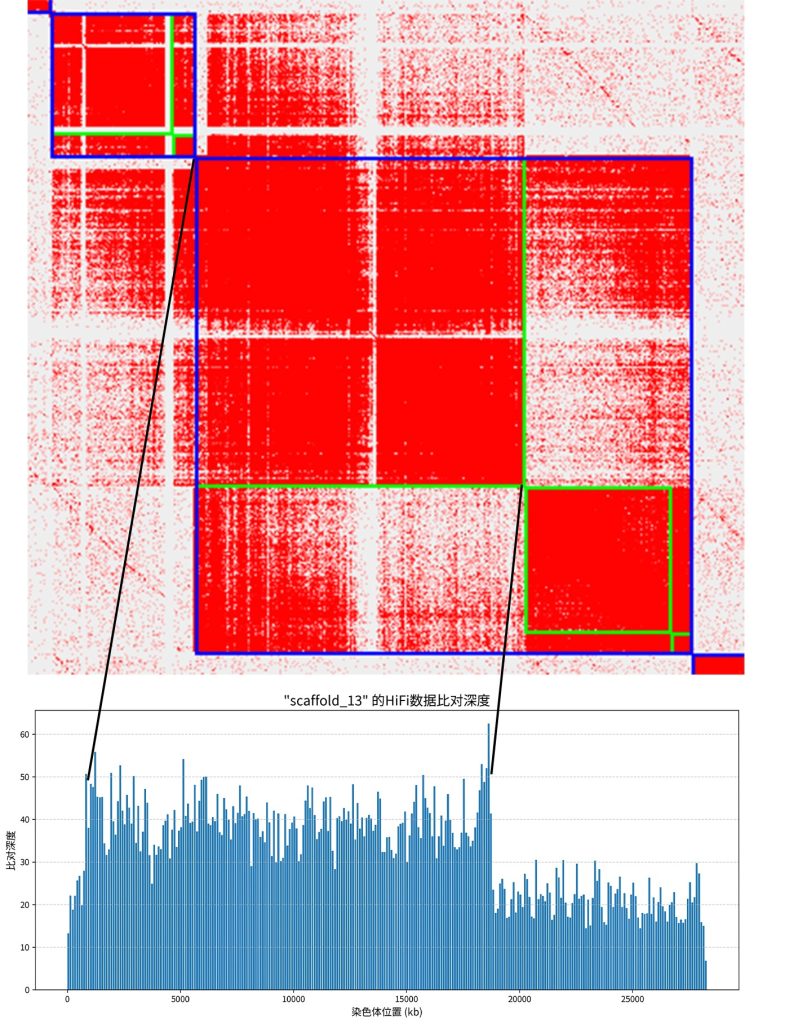
可以看到有一个非常明显的collpase区域,深度超过了40。这些区域可以和HIC热图上的collpase区相互对应。接下来只要找到这些contig就可以了。Juicebox非常好用,选中collapse区的contig就可以显示出这些contig的名字,整理到一个文件后,然后根据out_JBAT.liftover.agp这个文件,就可以转换为原始的contigID。然后在把复制的和其他合并到一个文件,重新挂载就可以。
grep -f Copy_contig.txt out_JBAT.liftover.agp > Copy_contig_id.txt
seqkit grep -f Copy_contig_id.txt > Copy_contig.fasta
cat ../../out_JBAT.FINAL.fa Copy_contig.fasta > Addcopy_out_JBAT.FINAL.fa
bwa mem -5SP -t 30 Add_copy_out_JBAT.FINAL.fa ../../../../../../Hi-C/NY-1_1.fq.gz ../../../../../../Hi-C/NY-1_2.fq.gz | samblaster | samtools view - -@ 30 -S -h -b -F 3340 -o Add_copy_out_JBAT.FINAL.bam
~/Software/HapHiC/utils/filter_bam Add_copy_out_JBAT.FINAL.bam 1 --nm 3 --threads 50 | samtools view - -b -@ 50 -o Add_copy_out_JBAT.FINAL_filtered.bam
~/Software/HapHiC/haphic pipeline Add_copy_out_JBAT.FINAL.fa ./Add_copy_out_JBAT.FINAL_filtered.bam 60 --correct_nrounds 2 --threads 40 --processes 40 --outdir Add_copy_out_JBAT.FINAL_filter
2. 20251016加笔
距离上次更新这篇博客已经过去一个多月了,上次的策略是行不通的。原因是复制了坍塌区的序列后,HIC数据比对的时候,会出现重复比对,在后续过滤的时候会被直接过滤掉,导致坍塌区完全没有比对,因此没有信号。正确的思路是使用C-pasing这个软件中的Collapse功能,对复制坍塌区后对坍塌区的hic信号进行随机分配,确保坍塌区有信号。软件的作者主要是通过2种方法来识别坍塌区,分别是hifiasm的gfa文件和Mapping的depth,但是我测试下来,作者官网的流程只能应对2条染色体之间的坍塌区,如果某一坍塌区,存在3个及3三个以上染色体的坍塌,无法出来。经过本人的不懈努力,终于解决了这个问题。因此下面的教程分为两个板块(1)两个染色体的坍塌区的处理(2)三个及以上染色体坍塌区的处理
2.1 两个染色体坍塌区
两个染色体之间的坍塌区相对简单,识别到坍塌区后,直接把坍塌的contig复制,然后分配clutster,重命名,重新挂载分配信号就可以了。
# C-phasing挂载
cphasing pipeline -f hap_n_12.hic.p_utg.fa -hic1 ../../../../Hi-C/NY-1_1.fq.gz -hic2 ../../../../Hi-C/NY-1_2.fq.gz -t 40 -n 8:8
# -hic1 -hic2 双端的测序文件
# -n 8:8 这个指的是“倍性:染色体数量”,例如,16倍体,倍性为8,那么一个单倍型就是8:8
# 基于Hifiasm生成的gfa文件识别坍塌区
cphasing collapse from-gfa ../hap_n_12.hic.p_utg.noseq.gfa
# 生成一个contigs.collapsed.contig.list,这是塌陷的contig
# 将坍塌的contig进行分配到相应的clusters(染色体)
phasing collapse rescue 3.hyperpartition/NY-1.pairs.q1.e5m.hg hap_n_12.hic.p_utg.contigsizes 3.hyperpartition/output.clusters.txt contigs.collapsed.contig.list -n 4 -at 3.hyperpartition/hap_n_12.hic.p_utg.allele.table
# 生成一个collapsed.rescue.clusters.txt和collapsed.rescue.contigs.list,记录坍塌contig的分配和重命名的规则
# 重新运行Scaffolding
cphasing scaffolding collapsed.rescue.clusters.txt ./2.prepare/NY-1.counts_AAGCTT.txt \
./2.prepare/NY-1.clm.gz \
-at ./3.hyperpartition/hap_n_12.hic.p_utg.allele.table \
-sc ./2.prepare/NY-1.split.contacts \
-f ../hap_n_12.hic.p_utg.fa \
-t 40 \
-o groups.agp \
-m precision
# 生成一个新的agp文件,这时新复制的坍塌contig已经挂在成功并且分配到了信号,但是新复制的contig ID和之前的是重复的
# 生成一个新的contig水平的fasta和agp文件,主要目的是重命名复制的contig
cphasing agp2fasta groups.agp ../hap_n_12.hic.p_utg.fa --contigs > draft.dup.fasta
cphasing collapse agp-dup groups.agp > groups.dup.agp
# 生成新的pairs.pqs文件
cphasing collapse pairs-dup NY-1.pairs.pqs collapsed.rescue.contigs.list -o sample.dup.pairs.pqs
# 生成Juicebox的输入文件.assmbly
cphasing utils agp2assembly groups.dup.agp > groups.dup.assembly
cphasing pairs2mnd sample.dup.pairs.pqs -o sample.dup.mnd.txt
# 生成Juicebox的输入文件.hic,注意,这一步需要用到3d-dna,请自行下载,并找到run-assembly-visualizer.sh文件
bash ~/Software/3d-dna/visualize/run-assembly-visualizer.sh groups.dup.assembly sample.dup.mnd.txt
# 生成新的corrected.pairs.gz(可选步骤,只有对contig进行切割后才需要)
cphasing-rs pairs-break sample.dup.pairs.pqs break.bed
# 基于调整的groups.review.assembly生成最终的agp文件
cphasing utils assembly2agp groups.dup.review.assembly -n 8:8
# 这一步会生成groups.dup.review.agp和groups.dup.review.corrected.agp(只有contig进行切割后才会生成,并且后续分析都要使用这个文件替代groups.dup.review.agp)
# 生成最后的基因组
cphasing agp2fasta groups.review.agp draft.asm.fasta > groups.review.asm.fasta
# 绘制新的热图
cphasing pairs2cool sample.dup.pairs.pqs sample.dup.pairs.pqs/_contigsizes sample.10k.cool
cphasing plot -a groups.dup.review.agp -m sample.10k.cool -o sample.500k.wg.png
2.1 三个及三个以上染色体坍塌区
这种情况下相对复杂,主要问题出在生成“collapsed.rescue.clusters.txt”文件的步骤,因为存在坍塌区在重新分配的时候,出现了多种情况,所以会出现只分配到三个染色体中的两个,或者直接不分配。这种情况下解决方法其实也很简单,就是手动修改“collapsed.rescue.clusters.txt”文件,将坍塌区的contiguous进行手动的分配。随后手动修改“collapsed.rescue.contigs.list”文件,给坍塌区的contig进行多次重命名,然后衔接下游的流程即可。
# C-phasing挂载
cphasing pipeline -f hap_n_12.hic.p_utg.fa -hic1 ../../../../Hi-C/NY-1_1.fq.gz -hic2 ../../../../Hi-C/NY-1_2.fq.gz -t 40 -n 8:8
# -hic1 -hic2 双端的测序文件
# -n 8:8 这个指的是“倍性:染色体数量”,例如,16倍体,倍性为8,那么一个单倍型就是8:8
# 基于Hifiasm生成的gfa文件识别坍塌区
cphasing collapse from-gfa ../hap_n_12.hic.p_utg.noseq.gfa
# 生成一个contigs.collapsed.contig.list,这是塌陷的contig
# 将坍塌的contig进行分配到相应的clusters(染色体)
phasing collapse rescue 3.hyperpartition/NY-1.pairs.q1.e5m.hg hap_n_12.hic.p_utg.contigsizes 3.hyperpartition/output.clusters.txt contigs.collapsed.contig.list -n 4 -at 3.hyperpartition/hap_n_12.hic.p_utg.allele.table
# 生成一个collapsed.rescue.clusters.txt和collapsed.rescue.contigs.list,记录坍塌contig的分配和重命名的规则
接下来,手动修改“collapsed.rescue.clusters.txt”分配坍塌区的contig
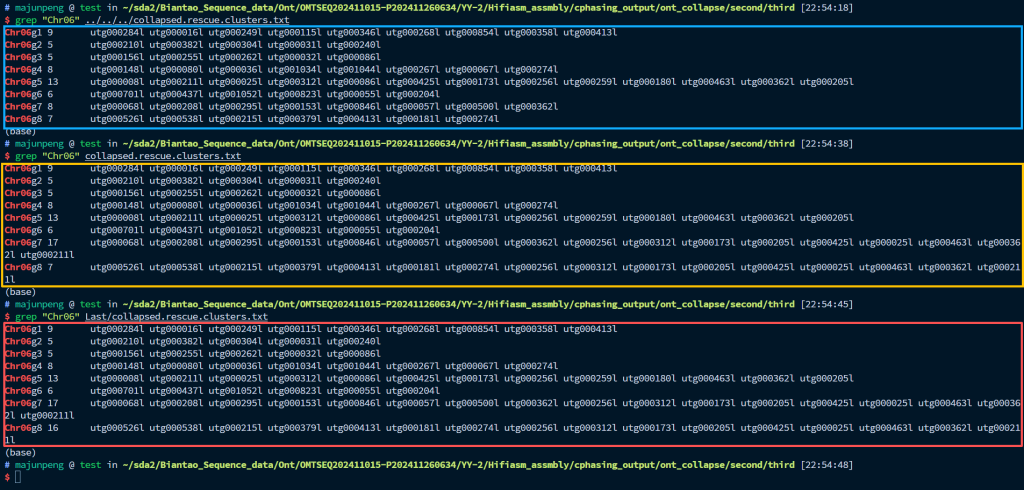
如图所示,蓝色框代表的是直接分配后的结果,可以看到Chr06g7和Chr06g8没有得到任何分配,因此要手动进行分配。但是怎么知道这些坍塌区有那些contig呢?其实很简单,直接把初始的挂载结果导入进juicebox,选中坍塌区,就能看到了。然后把这些contig,分别添加到Chr06g7和Chr06g8中,就可以了。黄色框为添加到了其中一个染色体,红色框为两个都添加。这一步之后,我们还要手动修改“collapsed.rescue.contigs.list”文件,对坍塌区中的contig进行手动的重命名。
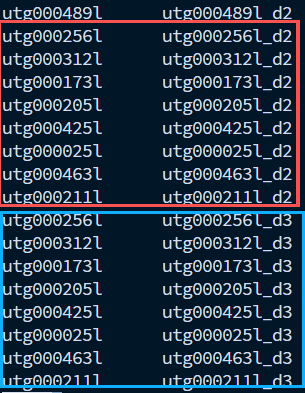
软件的默认命名规则为“原始的contigid_d2”,那么如果如果有多个拷贝,就是_d3,_d4逐渐增加。因此,手动改成d_3。这样我们就拿到了正确分配contig的collapsed.rescue.clusters.txt和collapsed.rescue.contig.list,衔接下面的步骤即可。因为后续和两条染色体的下游相同,就不写了,参考上面的即可。
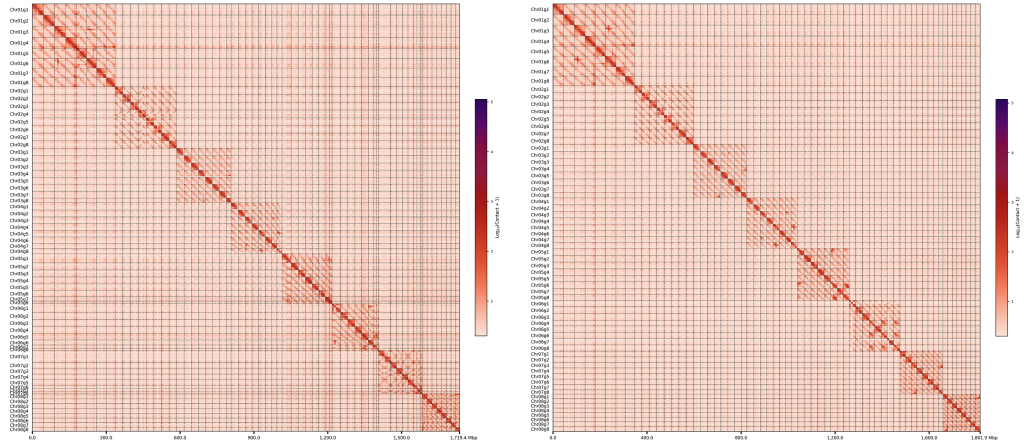
左边是未处理坍塌区的hic热图,右边是处理后的,可以看到,坍塌区明显被分成了多个,并且处理前的基因组大小为1719.4,处理后为1801.9,说明坍塌区的成功复制,导致基因组变大了。