WGDI: 全基因组加倍(WGD)
1.数据准备和预处理
WGDI要求(1)基因组文件;(2)基因组注释文件(gff);(3)CDS;(4)蛋白序列
# WGDI 对基因组注释、CDS和蛋白序列文件有独特的格式和ID要求,可以通过作者的脚本来进行转换,脚本下载地址,
python ./Script/getgff.py Genome.gff tmp_Genome.gff
# Genome.gff:基因组注释文件
# tmp_Genome.gff:转换的中间过程文件,后续会用到
python ../Script/gff_lens.py tmp_Genome.gff WGDI_Genome.gff Genome_length.txt
# WGDI_Genome.gff:WGDI要求的特定格式的基因组注释文件
# Genome_length.txt:基因组信息统计结果,统计了每条染色体的长度和基因的数量
python ../Script/seq_newname.py WGDI_Genome.gff Genome_pep.fasta WGDI_Genome_pep.fasta
# Genome_pep.fasta:蛋白文件
# WGDI_Genome_pep.fasta:根据新生成的gff文件,重命名后的蛋白文件
python ../Script/seq_newname.py WGDI_Genome.gff Genome_cds.fasta WGDI_Genome_cds.fasta
# Genome_pep.fasta:CDS文件
# WGDI_Genome_pep.fasta:根据新生成的gff文件,重命名后的CDS文件
2. 运行WGDI
WGDI的运行为多模块分段,需要依次进行各个模块的运行。
# 运行 Blast
blastp -num_threads 150 -db Genome_pep.fasta -query Genome_pep.fasta -outfmt 6 -evalue 1e-5 -out Genme_blast.txt
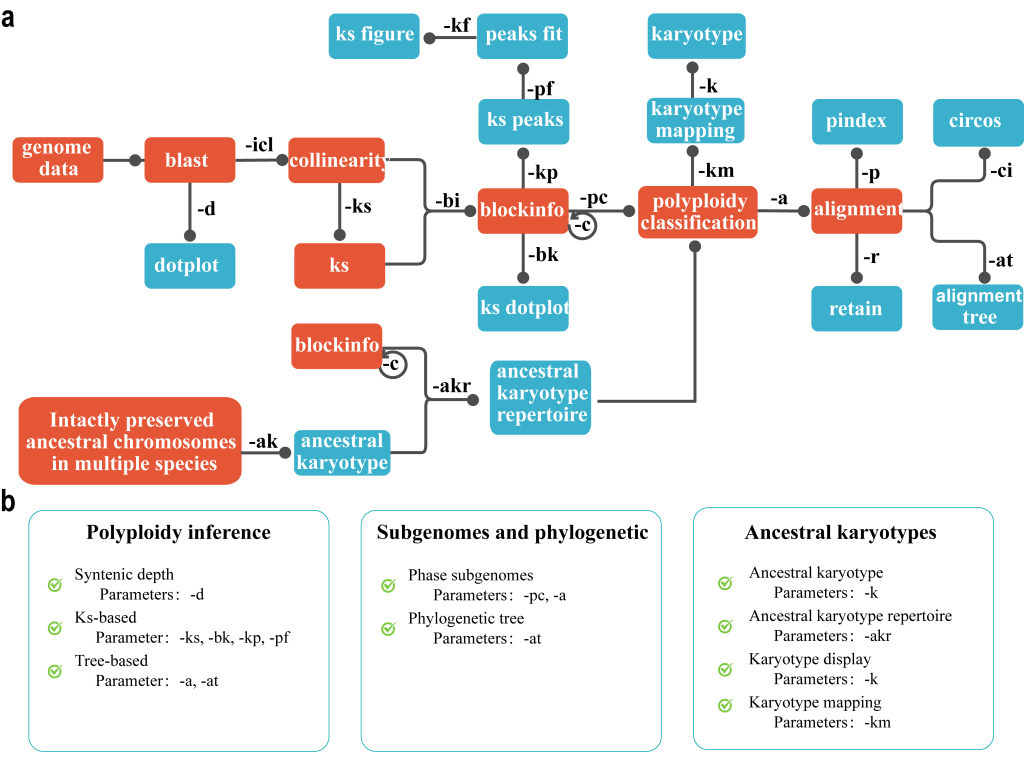
2.1 Collinearity 模块
# 生成配置文件
wgdi -icl \? >> Collinearity.conf ###正常是wgdi -icl ? >> Collinearity.conf,但是由于?需要转译,所以要在前面加一个\
# Collinearity 模块配置文件
[collinearity]
gff1 = gff1 file #基因组注释文件
gff2 = gff2 file #基因组注释文件
lens1 = lens1 file #基因组统计信息文件
lens2 = lens2 file #基因组统计信息文件
blast = blast file # Blast结果文件
blast_reverse = false
multiple = 1
process = 8
evalue = 1e-5
score = 100
grading = 50,40,25
mg = 40,40
pvalue = 0.2
repeat_number = 10
positon = order
savefile = Genome_collinearity.txt # 输出的结果文件
# 运行
wgdi -icl Collinearity.conf
2.2 Non-synonymous (Ka) and synonymous (Ks) 模块
# 生成配置文件
wgdi -ks ? >> KaKs.conf
# Non-synonymous (Ka) and synonymous (Ks) 配置文件
[ks]
cds_file = WGDI_Genome_cds.fasta # 基因组CDS文件
pep_file = WGDI_Genome_pep.fasta # 基因组蛋白序列文件
align software = muscle
pairs_file = Genone_Collinearity.txt
ks_file = Genone_Ksks.txt # 输出结果文件
# 运行
wgdi -ks KaKs.conf
2.3 BlockInfo 模块
# 生成配置文件
wgdi -ks ? >> BlockInfo.conf
# BlockInfo 配置文件
[blockinfo]
blast = Genome_blast.txt
gff1 = WGDI_Genome.gff
gff2 = WGDI_Genome.gff
lens1 = Genome_length.txt
lens2 = Genome_length.txt
collinearity = Genome_Collinearity.txt
score = 100
evalue = 1e-5
repeat_number = 10
position = order
ks = Genome_ks.txt
ks_col = ks_NG86
savefile = Genome_block.csv # 输出结果文件
# 运行
wgdi -bi BlockInfo.conf
2.4 BlockKs 模块
# 生成配置文件
wgdi -ks ? >> Blockks.conf
# BlockInfo 配置文件
[blockks]
lens1 = Genome_length.txt
lens2 = Genome_length.txt
genome1_name = Genome.fasta # 基因组序列文件
genome2_name = Genome.fasta # 基因组序列文件
blockinfo = Genome_block.csv
pvalue = 0.05
tandem = true
tandem_length = 200
markersize = 1
area = 0,2
block_length = 5
figsize = 8,8
savefig = Genome_Blockks.pnf # 输出的结果文件
# 运行
wgdi -bk Blockks.conf
2.5 Kspeaks 模块
# 生成配置文件
wgdi -ks ? >> KsPeaks.conf
# 配置文件
[kspeaks]
blockinfo = Genome_block.csv
pvalue = 0.2
tandem = true
block_ length = 5
ks_area = 0,10
multiple = 1
homo = -1,1
fontsize = 9
area = 0,3
figsize = 10,6.18
savefig = Genome_Kspeaks.png # Ks分布图
savefile = Genome_Kspeaks.csv # 输出结果文件
# 运行
wgdi -kp KsPeaks.conf
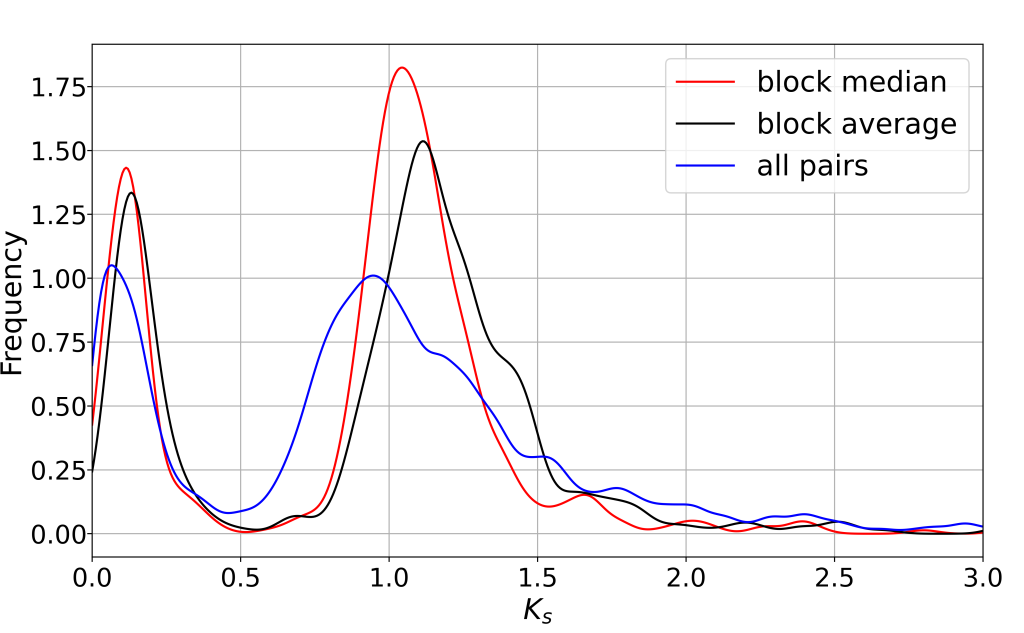
Ks分布图
Ks分布图可以看到两个明显的峰,Ks1(0.2)和Ks2(1.2)。WGDI的KsPeaks模块一次只能拟合一个峰值,所以我们要重新运行Kspeaks模块,分别运行两次,得到2个结果。
# Ks1 配置文件
[kspeaks]
blockinfo = Genome_block.csv
pvalue = 0.2
tandem = true
block_ length = 5
ks_area = 0,0.75
multiple = 1
homo = -1,1
fontsize = 9
area = 0,3
figsize = 10,6.18
savefig = Genome_Kspeaks_1.png # Ks1分布图
savefile = Genome_Kspeaks_1.csv # Ks1输出结果文件
# Ks1 配置文件
[kspeaks]
blockinfo = Genome_block.csv
pvalue = 0.2
tandem = true
block_ length = 5
ks_area = 0.75,2
multiple = 1
homo = -1,1
fontsize = 9
area = 0,3
figsize = 10,6.18
savefig = Genome_Kspeaks_2.png # Ks2 分布图
savefile = Genome_Kspeaks_2.csv # Ks2 输出结果文件
# 运行
wgdi -kp KsPeaks.conf
2.6 PeakFit 模块
# 生成配置文件
wgdi -ks ? >> PeakFit.conf
# 配置文件
[peaksfit]
blockinfo = Genome_Kspeaks_1.csv
mode = median
bins_number = 200
ks_area = 0,10
fontsize = 9
area = 0,3
figsize = 10,6.18
savefig = Genome_PeakFit_1.png
[peaksfit]
blockinfo = Genome_Kspeaks_2.csv
mode = median
bins_number = 200
ks_area = 0,10
fontsize = 9
area = 0,3
figsize = 10,6.18
savefig = Genome_PeakFit_2.png
# 运行
wgdi -pf PeakFit.conf
运行成功后,会打印曲线拟合的信息,后续的KsFigture模块会用到
2.7 KsFigure 模块
KsFigure 模块运行需要一个CSV格式的输入文件,格式如下,如果需要拟合多个曲线,可以依次增加行数。
| color | linewidth | linestyle | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Chr_Chr | green | 1 | -- | 5.02 | 0.83 | 0.10 | 2.08 | 1.83 | 0.25 |
# 生成配置文件
wgdi -ks ? >> KsFigure.conf
# 配置文件
[ksfigure]
ksfit = Genome_PeakFit.csv # PeakFit的拟合信息
labelfontsize = 15
legendfontsize = 15
xlabel = none
ylabel = none
title = none
area = 0,2
figsize = 10,6.18
shadow = true
savefig = Genome_Ks.png # Ks曲线拟合结果
# 运行
wgdi -kf KsFigure.conf
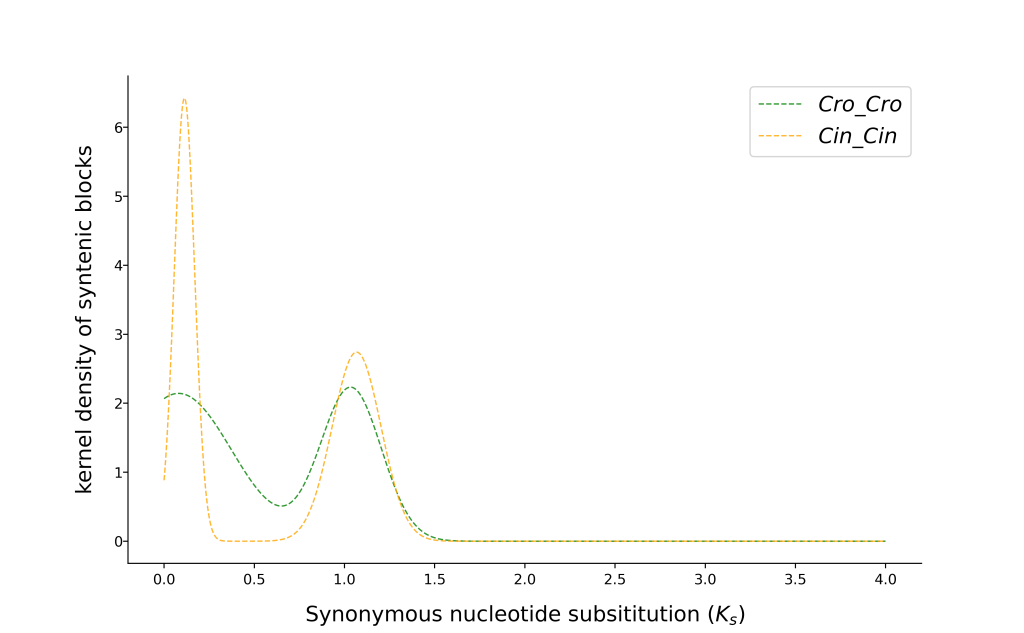
3. 循环运行
3.1 物种内Ks
Diamond 构建索引
for i in Oryza_sativa Olyra_latifolia Phyllostachys_edulis Guadua_angustifolia Dendrocalamus_latiflorus Bambusa_pervariabilis_Dendrocalamopsis_daii Bambusa_pervariabilis_Dendrocalamopsis_daii
do
diamond makedb --in ${i}.pep -d ${i}.pep
done
运行Diamond
for i in Oryza_sativa Olyra_latifolia Phyllostachys_edulis Guadua_angustifolia Dendrocalamus_latiflorus
do
diamond blastp -q ${i}.pep -d ${i}.pep -o ${i}_${i}.blast -p 40
done
批量生成WGDI配置文件
运行WGDI
for i in {Chrysanthemum_seticuspe,Chrysanthemum_indicum,Shidagonglao}
do
echo "
[collinearity]
gff1 = ${i}.gff
gff2 = ${i}.gff
lens1 = ${i}_length.txt
lens2 = ${i}_length.txt
blast = ${i}_${i}.blast
blast_reverse = no
multiple = 1
process = 50
evalue = 1e-5
score = 100
grading = 50,40,25
mg = 25,25
pvalue = 1
repeat_number = 10
positon = order
savefile = ${i}_${i}.icl
[ks]
cds_file = Conbine.cds
pep_file = Conbine.pep
align_software = muscle
pairs_file = ${i}_${i}.icl
ks_file = ${i}_${i}.ks
[blockinfo]
blast = ${i}_${i}.blast
gff1 = ${i}.gff
gff2 = ${i}.gff
lens1 = ${i}_length.txt
lens2 = ${i}_length.txt
collinearity = ${i}_${i}.icl
score = 100
evalue = 1e-5
repeat_number = 10
position = order
ks = ${i}_${i}.ks
ks_col = ks_NG86
savefile = ${i}_${i}_block.csv
[kspeaks]
blockinfo = ${i}_${i}_block.csv
pvalue = 0.2
tandem = true
block_length = 5
ks_area = 0,10
multiple = 1
homo = -1,1
fontsize = 9
area = 0,3
figsize = 10,6.18
savefig = ${i}_${i}_Kspeaks.png
savefile = ${i}_${i}_Kspeaks.csv
[peaksfit]
blockinfo = ${i}_${i}_block.csv
mode = median
bins_number = 200
ks_area = 0,10
fontsize = 9
area = 0,3
figsize = 10,6.18
shadow = true
savefig = ${i}_${i}_pf.png" > ${i}.conf
done
for i in Oryza_sativa Olyra_latifolia Phyllostachys_edulis Guadua_angustifolia Dendrocalamus_latiflorus Bambusa_pervariabilis_Dendrocalamopsis_daii
do
wgdi -icl ${i}.conf
wgdi -ks ${i}.conf
wgdi -bi ${i}.conf
wgdi -kp ${i}.conf
done
3.2 物种间Ks
运行Diamond
for i in Phyllostachys_edulis Oryza_sativa Olyra_latifolia Melocanna_humilis Guadua_angustifolia Dendrocalamus_latiflorus
do
diamond blastp -q ${i}.pep -d Bambusa_pervariabilis_Dendrocalamopsis_daii.pep -o ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.blast -p 40
done
批量生成WGDI配置文件
运行WGDI
for i in Phyllostachys_edulis Oryza_sativa Olyra_latifolia Melocanna_humilis Guadua_angustifolia Dendrocalamus_latiflorus
do
echo "
[collinearity]
gff1 = ${i}.gff
gff2 = Bambusa_pervariabilis_Dendrocalamopsis_daii.gff
lens1 = ${i}_length.txt
lens2 = Bambusa_pervariabilis_Dendrocalamopsis_daii_length.txt
blast = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.blast
blast_reverse = false
multiple = 1
process = 50
evalue = 1e-5
score = 100
grading = 50,40,25
mg = 25,25
pvalue = 1
repeat_number = 10
positon = order
savefile = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.icl
[ks]
cds_file = Conbine.cds
pep_file = Conbine.pep
align_software = muscle
pairs_file = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.icl
ks_file = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.ks
[blockinfo]
blast = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.blast
gff1 = ${i}.gff
gff2 = Bambusa_pervariabilis_Dendrocalamopsis_daii.gff
lens1 = ${i}_length.txt
lens2 = Bambusa_pervariabilis_Dendrocalamopsis_daii_length.txt
collinearity = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.icl
score = 100
evalue = 1e-5
repeat_number = 10
position = order
ks = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.ks
ks_col = ks_NG86
savefile = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii_block.csv
[correspondence]
blockinfo = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii_block.csv
lens1 = ${i}_length.txt
lens2 = Bambusa_pervariabilis_Dendrocalamopsis_daii_length.txt
tandem = true
tandem_length = 200
pvalue = 0.2
block_length = 5
tandem_ratio = 0.5
multiple = 1
homo = -1,1
savefile = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii_Correspondence.csv
[kspeaks]
blockinfo = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii_Correspondence.csv
pvalue = 0.2
tandem = true
block_length = 5
ks_area = 0,10
multiple = 1
homo = -1,1
fontsize = 9
area = 0,3
figsize = 10,6.18
savefig = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii_Kspeaks.png
savefile = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii_Kspeaks.csv
[peaksfit]
blockinfo = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii_Kspeaks.csv
mode = median
bins_number = 200
ks_area = 0,10
fontsize = 9
area = 0,3
figsize = 10,6.18
shadow = true
savefig = ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii_Peaksfit.png
" > ${i}_Bambusa_pervariabilis_Dendrocalamopsis_daii.conf
done
for i in Dendrocalamus_latiflorus_Bambusa_pervariabilis_Dendrocalamopsis_daii.conf Guadua_angustifolia_Bambusa_pervariabilis_Dendrocalamopsis_daii.conf Melocanna_humilis_Bambusa_pervariabilis_Dendrocalamopsis_daii.conf Olyra_latifolia_Bambusa_pervariabilis_Dendrocalamopsis_daii.conf Oryza_sativa_Bambusa_pervariabilis_Dendrocalamopsis_daii.conf Phyllostachys_edulis_Bambusa_pervariabilis_Dendrocalamopsis_daii.conf
do
wgdi -icl $i
wgdi -ks $i
wgdi -bi $i
wgdi -c $i
wgdi -kp $i
wgdi -pf $i
done