CAFE 5:基因家族的扩张与收缩
1. 软件下载
### 创建分析环境
mamba create -n Gene_family_expend_shrink
### 下载 cafe
mamba install bioconda::cafe
### 下载 orthofinder
2 基因家族聚类
### 运行orthofinder进行基因家族的聚类
orthofinder -f ./quanbu -o Orthofinder_result_20241128_pep_quanbu -a 30 -t 30
# -f 输入文件夹(包含所有待分析物种的蛋白序列的文件夹)
# -o 输出结果文件夹的名称
# -a Parallel sequence search 运行的线程数
# -t Paralle analysis 运行的的线程数
### 输出结果
Orthogroups/Orthogroups.GeneCount.tsv :后续分析的主要结果
3 基因家族的扩张与收缩分析
CAFE 的输入文件有两个,(1)符合二分叉的超度量树(带有分歧时间的系统发育树),(2)物种和基因家族数量的对应表。其中(1)应该是估算分歧时间后生成的结果,(2)可以通过Orthogroups/Orthogroups.GeneCount.tsv进行格式转换生成。
# Orthogroups.GeneCount.tsv转换生成Gene_family.txt
awk -v OFS="\t" '{$NF=null;print $1,$0}' Orthogroups.GeneCount.tsv |sed -E -e 's/Orthogroup/desc/' -e 's/_[^\t]+//g' > gene_families.txt
# 对基因家族进行过滤(可选步骤)
awk 'NR==1 || $3<100 && $4<100 && $5<100 && $6<100 && $7<100 && $8<100 && $9<100 && $10<100 && $11<100 && $12<100 && $13<100 && $14<100 && $15<100 && $16<100 && $17<100 {print $0}' gene_families.txt > gene_families_filter.txt
# 运行CAFE
cafe5 --infile gene_families.txt --tree Reroot_Complete_Time_point_tree.tre --cores 100 --output_prefix cafe_out
# --infile 物种和基因家族数量的对应表(1)
# --tree 超度量树(2)
# --cores 运行的线程数
# --output_prefix 输出文件的前缀
4 输出结果
# XXX_asr.tre
# XXX_branch_probabilities.tab
# XXX_change.tab
# XXX_clade_results.txt 最重要的结果,显示了进化树节点中扩张和收缩的基因
# XXX_count.tab
# XXX_family_likelihoods.txt
# XXX_family_results.txt
# XXX_report.cafe
# XXX_results.txt
5. 提取收缩和扩张的基因进行富集分析
5.1 提取扩张和收缩的基因基因
cat Gamma_change.tab |cut -f1,15|grep "+[1-9]" >sample.expanded #提取Gamma_change.tab第15列代表物种sample的扩张的orthogroupsID
cat Gamma_change.tab |cut -f1,15|grep "-" >sample.contracted #提取Gamma_change.tab第15列代表物种sample的收缩的orthogroupsID
grep "sample<13>\*" Gamma_asr.tre > sample_significant_trees.tre # 根据sample ID和编号提取sample分支的基因家族显著扩张或收缩的基因家族树(Gamma_asr.tre文件中默认以p<0.05为标准判断变化是否显著)
grep -E -o "OG[0-9]+" sample_significant_trees.tre > sample_significant.ogs # 提取sample分支显著变化的OG IDs (默认以p<0.05为标准)
awk '$2 <0.01 {print $1}' Gamma_family_results.txt >p0.01_significant.ogs # 以p<0.01为标准提取所有显著扩张或收缩的orthogroupsID(根据情况调整,常用p<0.05或p<0.01)
grep -f sample_significant.ogs p0.01_significant.ogs > sample_p0.01_significant.ogs # 提取以p<0.01为标准判断显著性的sample分支基因家族显著变化的OG IDs。
grep -f sample_p0.01_significant.ogs sample.expanded |cut -f1 >s ample.expanded.significant #提取显著扩张的sample物种的orthogroupsID
grep -f sample_p0.01_significant.ogs sample.contracted |cut -f1 > sample.contracted.significant #提取显著收缩的sample物种的orthogroupsID
grep -f sample.expanded.significant ./OrthoFinder/Results_Oct14/Orthogroups/Orthogroups.txt |sed "s/ /\n/g"|grep "bv" |sort -k 1.3n |uniq >sample.expanded.significant.genes #提取显著扩张的基因列表,假设基因ID是bv的前缀。
grep -f sample.contracted.significant ./OrthoFinder/Results_Oct14/Orthogroups/Orthogroups.txt sed "s/ /\n/g"|grep "bv" |sort -k 1.3n |uniq >sample.contracted.significant.genes #提取显著收缩的基因列表,假设基因ID是bv的前缀。
seqkit grep -f sample.expanded.significant.genes sample.pep.fa >sample.expanded.significant.pep.fa #提取显著扩张的基因序列
seqkit grep -f sample.contracted.significant.genes sample.pep.fa >sample.contracted.significant.pep.fa #提取显著收缩的基因序列
5.2 富集分析
富集分析要准备GO和KEGG注释的结果文件,文件的格式为第一列为基因ID,第二列为基因ID对应的KO或GO号。
# KEGG 注释结果文件格式
evm.TU.Chromosome_8_B.3367 K00001
evm.TU.Chromosome_4_B.3962 K00001
evm.TU.Chromosome_3_B.5527 K00001
evm.TU.Chromosome_8_B.3356 K00001
evm.TU.Chromosome_4_B.2409 K00001
evm.TU.Chromosome_8_B.3357 K00001
evm.TU.Chromosome_3_B.1839 K00001
evm.TU.Chromosome_4_B.2190 K00001
# GO注释结果文件格式
evm.model.Chromosome_3_B.1356 GO:0006979
evm.model.Chromosome_3_B.1367 GO:0010492
evm.model.Chromosome_4_B.331 GO:0006355
evm.model.Chromosome_9_B.2428 GO:0005634
evm.model.Chromosome_8_B.806 GO:0004497
evm.model.Chromosome_4_B.2710 GO:0003735
evm.model.Chromosome_5_B.3513 GO:0003729
GO和KEGG的富集分析使用TBtools完成,教程可以参考TBtools的官方教程
GO富集教程:https://geekdaxue.co/read/cjchen@hirv8i/wnve76
KEGG富集教程:https://geekdaxue.co/read/cjchen@hirv8i/pohup4
富集分析需要用到的数据库可以通过TBtools软件下载,但是可能会因为网络问题下载失败,我把它存在了百度网盘中,需要请自取:https://pan.baidu.com/s/1TvWrsp57FT9QT8H3Vf0gig
绘制GO和KEGG的富集图
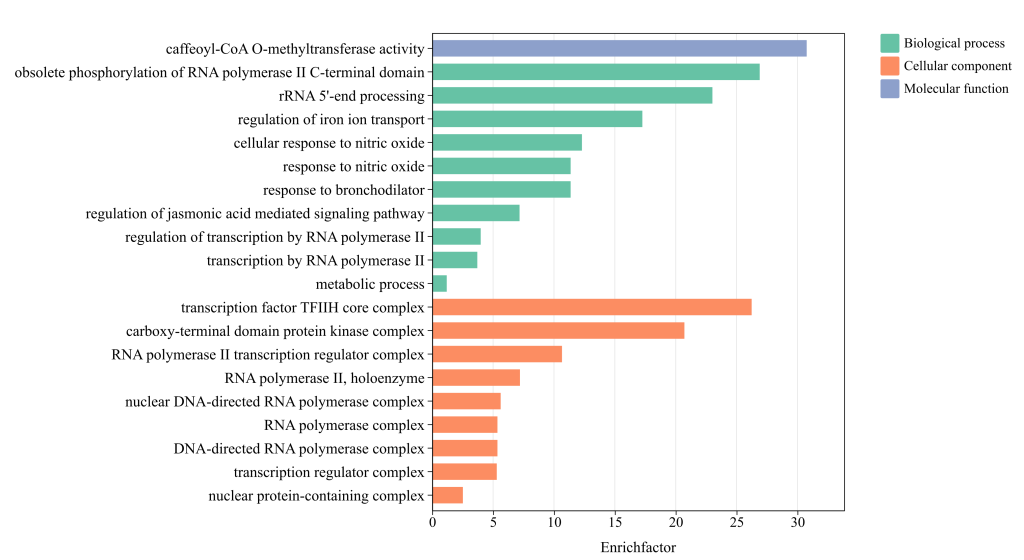
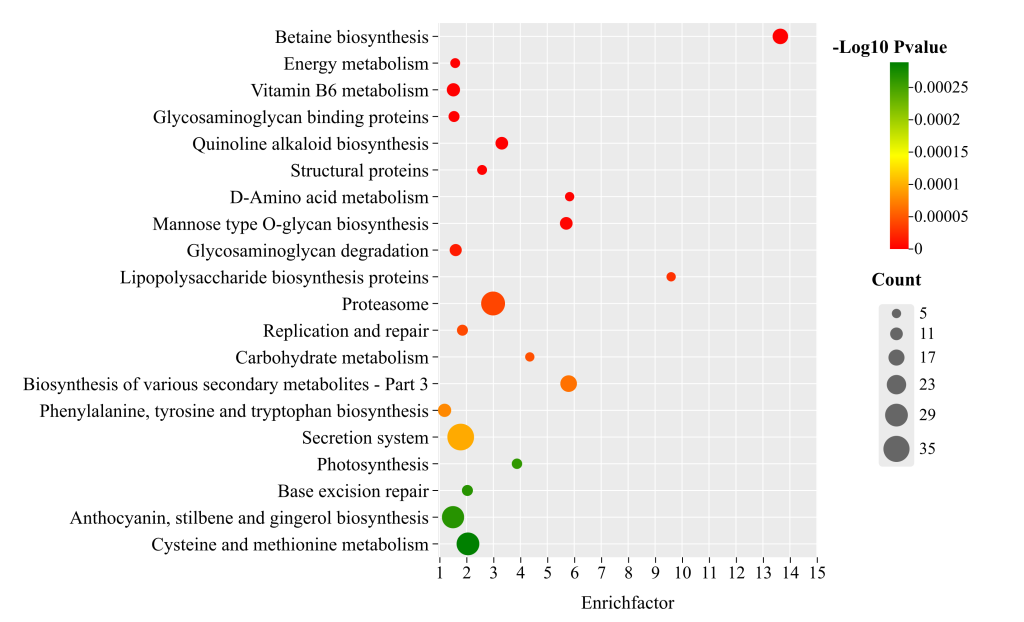
通常KEGG和GO会取前20个条目(P value or Count or Enrichfator)进行绘制,GO绘制为分类柱状图,KEGG绘制为气泡图,推荐使用Chiplot网站进行绘图,对0基础的生信小白很友好。
气泡图(KEGG富集)绘图网站:https://www.chiplot.online/enrich_plot.html
气泡图(KEGG富集)绘图网站使用教程:https://www.bilibili.com/video/BV1mP4y1w74C/?vd_source=ac0301f422193d385c811b0bdbbd27ff
分类柱状图(GO富集)绘图网站:https://www.chiplot.online/bar_plot_width_category.html
分类柱状图(GO富集)绘图网站使用教程:https://www.bilibili.com/video/BV1CU4y1w7WM/